Home
Raphaël Robiette
Research
People
Publications
Teaching
Contact Details
Links
The Robiette Research Group
Research in Organic and Physical Organic Chemistry
Physical Organic Studies
The fundamental understanding of the mechanism and factors governing stereoselectivity in the different processes we are interested in is crucial to the success of their development. Accordingly, one of our objective is to predict chemical behaviour (mechanism, reactivity, selectivity,...) in organic chemistry by combining experimental efforts (kinetic studies, KIE, crossover experiments, substituent effect studies,...) with computational methods (DFT, ab initio). The results of these studies enable us not only to improve these processes but also feed our imagination to design new methodologies.
This page lists some of our work in this area.
Imidazole-containing macrocycles
Mechanism and origin of E/Z selectivity in the modified Julia olefination
Our previous studies on olefination reactions (see for instance JACS 2006, 2394) led us to be concerned about the modified Julia olefination. This reaction consists in the preparation of alkenes from benzothiazol-2-yl (BT) sulfones and aldehydes. It has emerged these last years as a powerful tool for carbon-carbon double bond formation, in particular when two complex molecular fragments must be connected. The only shortcoming of this reaction is the difficulty of predicting and controlling stereoselectivity of the newly formed double bond (see for instance JOC 2012, 6358). Although the global mechanistic sequence depicted below is known and widely accepted a detailed atomistic account of the mechanism and selectivity of this reaction is still lacking.
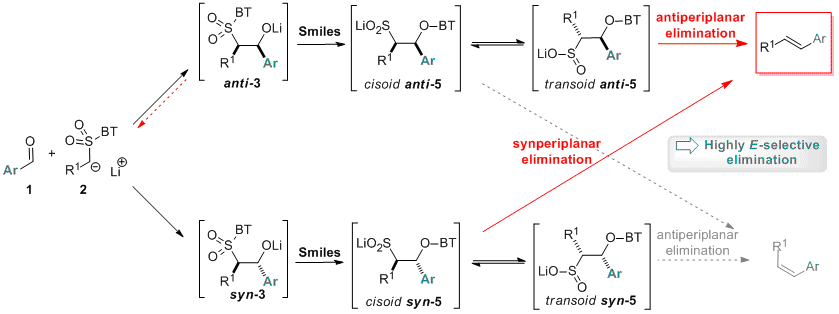
In collaboration with J. Pospíil, we use computational and experimental means to gain insights into the mechanism and factors controlling the E/Z selectivity in the modified Julia olefination of BT sulfones (see Eur. J. Org. Chem. 2013, 836). One of our main findings is that, in reaction of aromatic aldehydes, elimination occurs in fact through a concerted antiperiplanar and synperiplanar mechanism in the case of anti- and syn-sulfinate, respectively. Both diastereomeric pathways lead thus to the (E)-alkene.
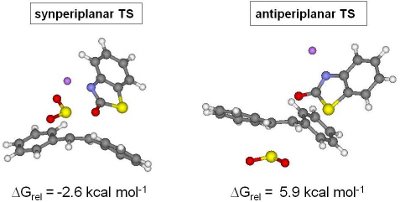
Transition state structure for synperiplanar and antiperiplanar elimination from syn-5 are depicted below.
This analysis now assists in the design of new reagents and reaction conditions and allows further development of modified Julia reaction for highly E/Z selective synthesis of alkenes.
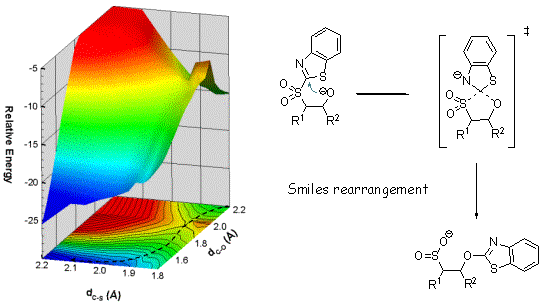
It is worth mentionning that, beside this clarification of the origin of E/Z selectivity, our study showed also that Smiles rearrangement, allowing the transfer of the BT group from sulfur to oxygen, occurs actually in a single elementary step (concerted process), in contrast to what is generally accepted. The potential energy surface around the TS is depicted below.

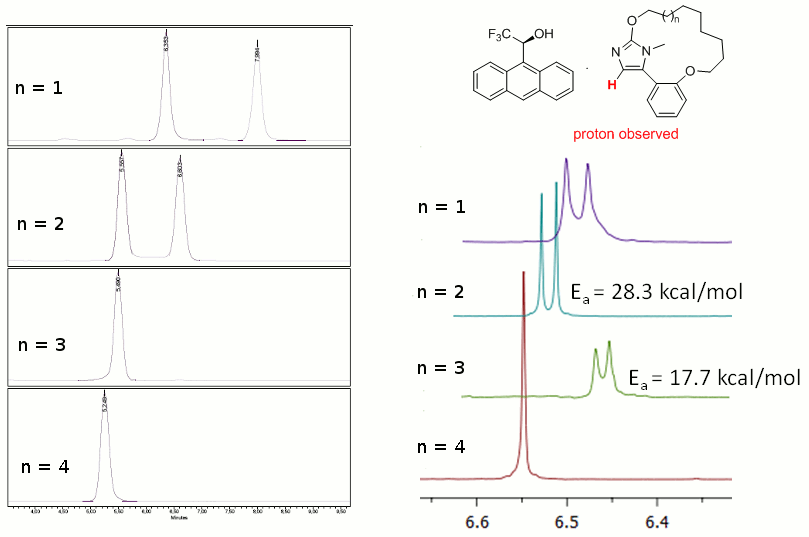
Chiral properties of imidazole-containing macrocycles
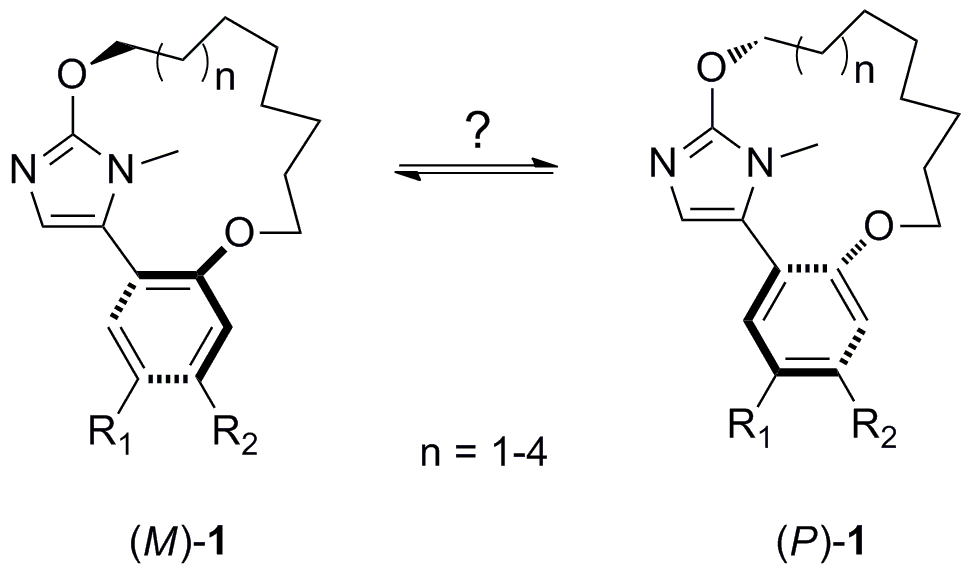
In the context of a medicinal chemistry project, we have designed and synthesized a new family of compounds comprising a 5-aryl-1H-imidazole motif included in a macrocycle (see the figure below). We reasoned that these macrocycles can potentially be chiral. Indeed, both their biaryl like and cyclophane-type motifs may well be a source of chirality. We thus decided to confirm the chiral properties of these macrocycles and investigate the barrier to stereoisomerisation (see Eur. J. Org. Chem. 2011, 6649).
Chiral HPLC and NMR studies in the presence of a chiral shift reagent showed that the two shorter macrocycles (n = 1, 2) are chiral whereas their superior homologues (n = 3, 4) isomerise rapidly at room temperature. Performing NMR analysis at different temperatures allowed determining the energy barrier to stereoisomerisation for macrocycles where n = 2 and 3.
In order to investigate further the origin of chirality in the case of shorter marcocycles (n = 1, 2) and the factors influencing the kinetic of enantiomerisation, we have optimised our macrocycles and explored the conformational equilibrium between the two atropisomers ((M)-1 and (P)-1). Computed energy barriers were in good agreement with our experimental observations and confirmed the significant decrease of the energy barrier to isomerisation with the size of the macrocycle. Our calculations gave also insights into the origin of the chirality: that is the cyclophane-type structure of macrocycles which is responsible for their chirality (and not their biaryl-type nature). Our computational results showed moreover that the mechanism of isomerisation involves a rope-skipping motion of the alicyclic chain going through the imidazole plane on the C4-H side and not on the N-Me side.
Reactions of sulfonium ylides
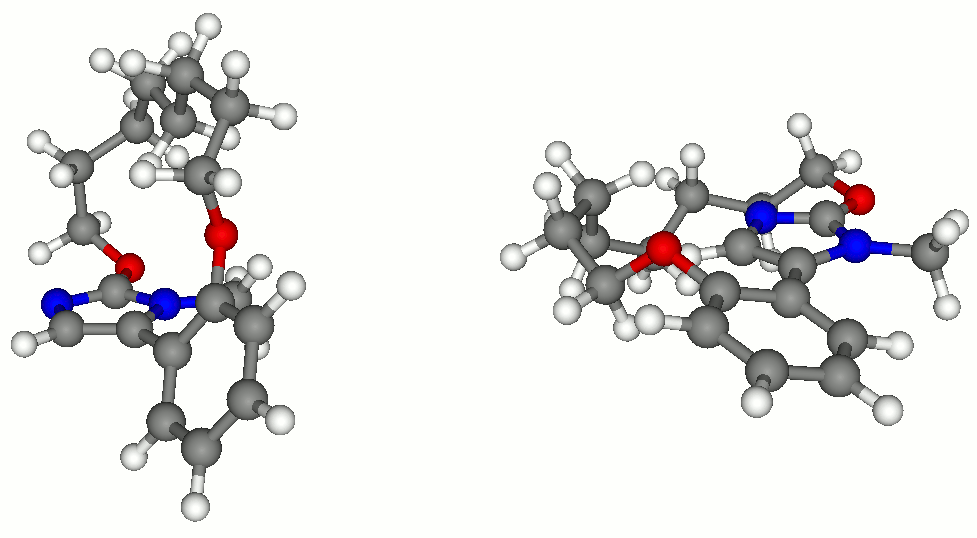
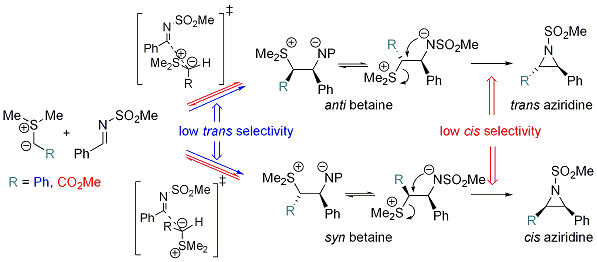
In one piece of work, we investigated the reaction of aziridine formation from sulfonium ylides and imines (J. Org. Chem. 2006, 71, 2726). This study enabled us to get a clear understanding of the mechanism of the process and to identify the origin of diastreoselectivity, and how this latter changes with stabilization of the ylide (R = Ph or CO2Me).
The pictures below show the two diasteromeric TSs for the addition of a semi-stabilized sulfur ylide (R = Ph) to a N-sulfonyl imine. The low selectivity in favor of the TS on the left, leading to trans aziridine, is accounted for by the favorable Coulombic interactions and stabilization by CH-O hydrogen bonding.
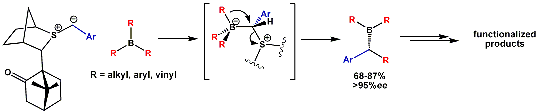
In collaboration with Profs Varinder Aggarwal and Jeremy Harvey, we are also considering the reaction of sulfur ylides with organoboranes. This methodology has been developed in the lab by G. Fang (see J. Am. Chem. Soc. 2005, 127, 1642 and Angew. Chem. 2007, 46, 359).
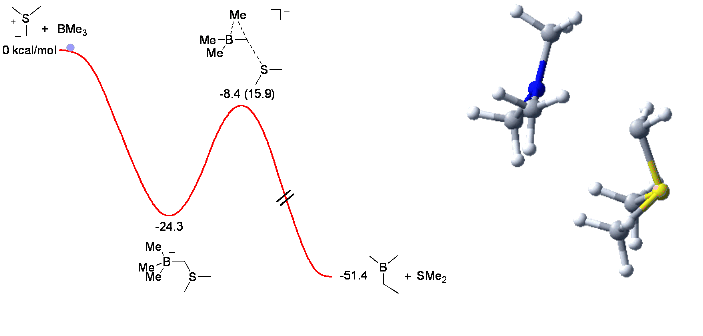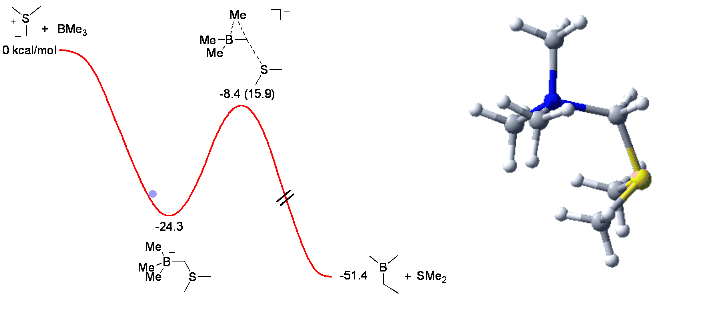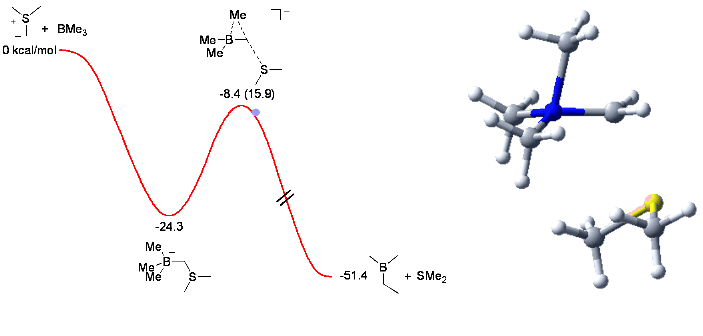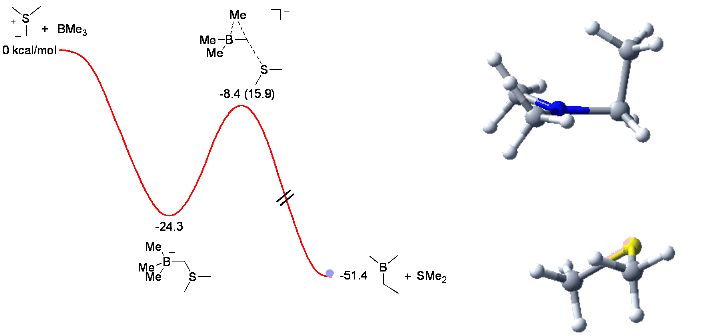
When trying the reaction with mixed organoboranes (BR1R2R3), we observed an unexpected and dramatic difference in relative migratory aptitude compared to those reported for similar rearrangements (Pinacol, Wagner-Meerwein,...). In order to understand this discrepency, we investigated the reaction by computational means. This enabled us to understand and identify the factors controlling the observed selectivity (see Chem. Comm. 2006, 741) with these results serving us now to design new mixed boranes.
The animation below shows the reaction of BMe3 with Me2S-CH2 leading to the corresponding homologated borane (Me2BEt) and SMe2.
We are also now using computational means to try to shed light on unexpected substituent effects we observed with ylides other than benzilic ones (see for instance Org. Biomol. Chem. 2008, 1185).
Chiral properties of tetrathiatriarylmethyl spin probes
Stable tetrathiatriarylmethyl radicals 1 belong to a family of trityl radicals which are extensively used for electron paramagnetic resonnance imaging (EPRI), oximetry, dynamic nuclear polarization ((DNP) and the detection of superoxide radical anion. These paramagnetic species have indeed unique properties: a high water solubility, an exceptional biostability, a narrow EPR linewidth and a low concentration-dependant broadning of the EPR line.
Surprisingly, during the numerous developments of trityl radicals for biomedical use, the fundamental question of potential chirality associated with their helicoidal conformation has never been addressed. It is however known that trityl radicals adopt a propeller shape in which all rings have the same diretion of twist (see the figure below). This geometrical arrangement allows two conformations, namely the right-handed (P) and the left-handed (M) helices, which feature an enantiomeric relationship.
Using experimental (HPLC, NMR and kinetic studies) and computational methods, we have been able to identify the mechanism of enantio-isomerisation for these tetrathiatriarylmethyl radicals and determine the corresponding energy barrier (see Chem. Comm. 2011, 4793 and Eur. J. Org. Chem. 2012, 6517). This enabled us to show that these radicals are chiral molecules at room temperature and that the two enantiomers that differ in their helicity are configurationally stable enough to be separated ans stored independantly for months.

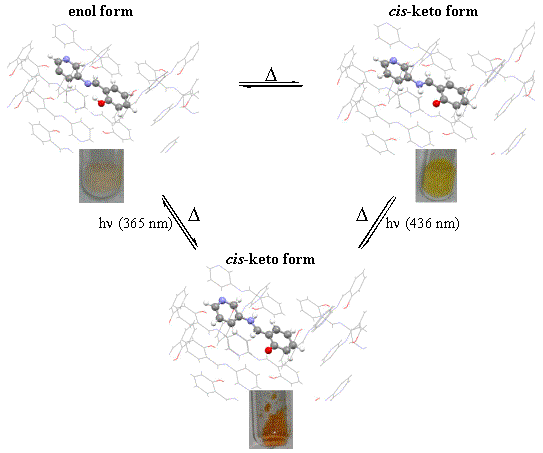
Photochromism and thermochoromism of N-salicylidene aminopyridines
In the context of a collaboration with the laboratory of molecular electronic of Professor Y. Garcia (UCL), we are interested in getting a better understanding of the mechanisms involved in the thermo- and photochromic properties of N-salicylidene aminopyridines in the crystalline-state. The first results of this study led us to reassess in depth the hypothesis previously stated concerning the optical properties of these molecules in the crystalline-state (see Chem. Eur.J. 2009, 4327).
This page and all its
contents belong to and were written by Raphaël Robiette.
For any comments or suggestions, please contact me.