Lysosomes
« It is obvious that no great advance can be made in the study of cell structure unless we succeed in separating the various cell constituents and obtain them in quantities sufficient to permit the application of the usual analytical methods.
Albert Claude (1941)
Glucose 6-phosphatase
L’insuline est une hormone secrétée par les cellules β des îlots de Langherans du pancréas ; elle participe au maintien dans des limites étroites de la concentration sanguine en glucose : lorsque la glycémie augmente au-delà d’une limite supérieure, le pancréas déverse de l’insuline dans le courant sanguin. L’hormone se fixe sur un récepteur de la surface des cellules hépatiques ; celles-ci captent des molécules de glucose dans la circulation sanguine et les stockent sous forme de glycogène, un polymère de glucose qui s’accumule en « rosettes » dans le cytosol des hépatocytes. Lorsque l’organisme a besoin d’énergie, des molécules de glucose sont détachées du glycogène et la glycogène phosphorylase transforme le glucose en glucose 1-phosphate puis en glucose 6-phosphate. La glucose 6-phosphatase hydrolyse cet ester phosphate et libère du glucose qui passe dans la circulation sanguine :
![]()
L’existence de cet enzyme fut découverte par Gerty T. Cori, Carl F. Cori et Gerhard Schmidt G (Department of Pharmacology, Washington University School of Medicine, St Louis). Henri Géry Hers (Laboratoire de chimie physiologique, Université Catholique de Louvain) entreprit sa purification à partir de foie de rat ; après précipitation à pH 5, il était impossible de remettre l’enzyme en solution. Dans son article de 1946, dans The Journal of Experimental Medicine, Albert Claude (Rockefeller Institute for Medical Research) insistait sur le fait que les organites subcellulaires s’agglutinent de manière irréversible à pH 5 ; il se forme des agrégats si le milieu utilisé pour homogénéiser les tissus n’est pas ajusté à un pH voisin de la neutralité. Christian de Duve en déduisit que la glucose 6-phosphatase est associée à un organite subcellulaire. La façon de le vérifier était de recourir au fractionnement subcellulaire. Jacques Berthet adapta la technique de Claude au fractionnement d’un homogénat de foie de rat, en prenant en compte les améliorations apportées par George Hogeboom, Walter Schneider et George Palade (Rockefeller Institute for Medical Research) : homogénéisation des tissus avec l’appareil en verre et piston en téflon de Potter et Elvejhem (Claude utilisait un mortier et un pilon) dans une solution de saccharose 250 millimolaire (Claude utilisait des solutions salines). Le protocole aboutissait à la séparation de trois fractions particulaires : fraction nucléaire, gros granules (Large Granules), petits granules (Small Granules) et d’un surnageant. La glucose-6 phosphatase sédimentait principalement dans la fraction des petits granules (microsomes), mais les fractions nucléaire et des gros granules (fraction mitochondriale) en contenaient aussi.
Références : Cori GT, Cori CF, Schmidt G The role of glucose-1-phosphate (1939)
Claude A Fractionation of mammalian liver cells by differential centrifugation (1946)
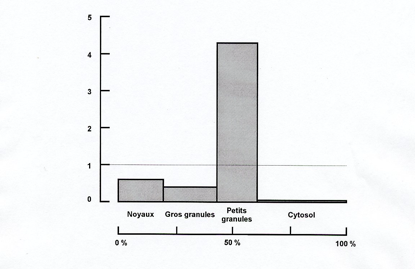 |
| La glucose 6-phosphatase est présente en proportions variables dans toutes les fractions particulaires. Ordonnée : activité spécifique relative ; abscisse : teneur en protéine (en % de l’homogénat) |
Ce résultat pouvait être interprété de deux façons : (i) les organites ont une composition enzymatique hétérogène ; il y a de la glucose 6-phosphatase en proportions variables dans les noyaux, les mitochondries et les microsomes, ceux-ci étant les plus riches en phosphatase ; (ii) les fractions subcellulaires sont hétérogènes ; la glucose 6-phosphatase n’est présente que dans les microsomes ; sa présence dans les fractions nucléaire et mitochondriale s’explique par l’existence d’une contamination par des microsomes. Il était logique de supposer que, pour qu’un organite remplisse une ou des fonctions spécifiques au sein de la cellule, il doit disposer d’un équipement enzymatique spécifique. Christian de Duve et Jacques Berthet optèrent pour cette interprétation et, à l’époque, ils furent parmi les premiers sinon les seuls à le faire. Le postulat de la localisation unique (un enzyme n’est présent que dans un seul type d’organite) allait jouer un rôle essentiel dans l’interprétation des résultats du fractionnement subcellulaire.
Ce point mérite qu’on s’y attarde. Dans une population d’organites comme celle des mitochondries, par exemple, les tailles varient : il y a de grosses mitochondries, des petites et, entre les deux, un éventail de mitochondries de taille moyenne. Il en va de même pour les lysosomes, dont la taille est en moyenne voisine de celle des mitochondries. Les populations d’organites sont « polydisperses ». La taille d’un organite déterminant sa vitesse de sédimentation, les fractions isolées sont des mélanges en proportions variables d’organites de taille semblable mais appartenant à des populations différentes, ce qui se traduit sur le plan biochimique par le fait que « les fractions contiennent un peu de tout ». De nombreux expérimentateurs commirent l’erreur de conclure que les enzymes de la fraction mitochondriale étaient tous présents dans les mitochondries. Au début des années 1950, la confusion entre fraction et organite conduisit des biochimistes à conclure que les chloroplastes contiennent de la peroxydase (un enzyme des peroxysomes). L’interprétation correcte du résultat de l’expérience représentée dans la figure ci-dessus est donc que la glucose 6-phosphatase est un enzyme microsomial et que les fractions nucléaire et mitochondriale sont contaminées par des microsomes en proportions variables. Les aspects théorique et pratique du fractionnement subcellulaire par centrifugation différentielle furent exposés dans l’article publié en 1954 par de Duve et Berthet dans International Review of Cytology.
Référence : De Duve C, Berthet J The use of differential centrifugation in the study of tissue enzymes (1954)
Phosphatase acide
La glucose-6 phosphatase fut le premier enzyme identifié dans les microsomes à une époque où le lien entre microsomes et réticulum endoplasmique n’était pas encore établi avec certitude. Sa distribution dans les fractions fut comparée à celle de la phosphatase acide, un enzyme hydrolysant aussi le glucose 6-phosphate mais à une vitesse moins élevée que la glucose-6 phosphatase. Pour le dosage de la phosphatase acide, le substrat utilisé était une molécule synthétique : le β-glycérophosphate. Nous savons aujourd’hui que le substrat in vivo de la phosphatase acide est un sucre phosphorylé : le mannose 6-phosphate. Les enzymes destinés aux lysosomes sont des glycoprotéines synthétisées dans le reticulum endoplasmique. Elles portent une chaîne glycanique se terminant par un mannose. Dans le cis-Golgi, ce mannose est phosphorylé. Dans le trans-Golgi, l’enzyme portant l’« étiquette mannose 6-phosphate » est reconnu par un récepteur spécifique. Les enzymes liés au récepteur sont séquestrés dans des vésicules qui se détachent du trans-Golgi et s’acidifient progressivement par fusion avec des endosomes. Au pH lysosomial (4,8), le mannose 6-phosphate se détache du récepteur et les enzymes lysosomiaux sont libérés. Le rôle de la phosphatase acide est d’hydrolyser la liaison ester phosphate du mannose 6-phosphate. La chaîne glycanique des glycoprotéines lysosomiales ne porte pas de mannose 6-phosphate.
Après fractionnement par centrifugation d’un homogénat de foie de rat, la phosphatase acide présentait une particularité : dans les fractions fraîchement préparées, son activité était faible ; dans les fractions conservées quelques jours au réfrigérateur, elle était dix fois plus élevée. Une partie de l’activité dans les fractions « fraîches » était masquée – phénomène qualifié de « latence enzymatique ». Les traitements altérant les membranes (vieillissement, congélation et décongélation, traitement mécanique, détergents) démasquaient l’activité. Christian de Duve formula l’hypothèse que la phosphatase acide était localisée à l’intérieur d’un organite entouré d’une membrane imperméable au substrat (β-glycérophosphate) et que l’imperméabilité de cette membrane était abolie par le vieillissement des fractions. Cet organite hypothétique semblait distinct des mitochondries, sans que l’on puisse exclure qu’il s’agisse de petites mitochondries, de mitochondries « légères » ou de fragments de mitochondries [1], [2] . Deux autres enzymes se comportaient comme la phosphatase acide (sédimentation dans la fraction mitochondriale, maximum d’activité à pH 5, phénomène de latence) : la β-glucuronidase (qui hydrolyse les esters d’acide glucuronique) et la cathepsine (une protéase).
Références : Berthet J, de Duve C Tissue Fractionation Studies. I. The existence of a mitochondria-linked, enzymatically inactive form of acid phosphatase in rat-liver tissues (1951)
de Duve C, Berthet J, Berthet L, Appelmans F Permeability of Mitochondria (1951)
A la suite d’une modification du protocole de fractionnement, la fraction des gros granules fut séparée en deux fractions appelées respectivement M (Mitochondria) et L (Light Mitochondria). Avec le schéma de fractionnement de Claude, la cytochrome oxydase sédimentait pour l’essentiel dans la fraction mitochondriale, la glucose 6-phosphatase dans les microsomes et la phosphatase acide occupait une position intermédiaire entre cytochrome oxydase et glucose 6-phosphatase. Après modification, le profil de distribution de la phosphatase acide changea radicalement : il présentait un pic dans la fraction L et se démarquait nettement des distributions de la cytochrome oxydase et de la glucose 6-phosphatase. L’hypothèse d’une association de la phosphatase acide à de petites mitochondries perdait de sa crédibilité ; on était plutôt en présence d’un organite de nature inconnue. En parcourant la littérature scientifique, Jacques Berthet dressa une liste d’enzymes présentant une distribution intermédiaire entre mitochondries et microsomes. Leur distribution dans les fractions N (Nuclei), M (Mitochondria), L (Light mitochondria), P (Particles), S (Supernatant) fut comparée à celle de la phosphatase acide. Deux nucléases montraient des profils de distribution et une latence comparables à ceux de la phosphatase acide, de la β-glucuronidase et de la cathepsine D. Ces enzymes étaient aussi des hydrolases. Le concept d’organite lytique ou lysosome prit naissance en 1955. Aux critères enzymatiques et physicochimiques, il fallait ajouter des critères morphologiques, c’est-à-dire « voir » les organites lytiques dans la cellule.
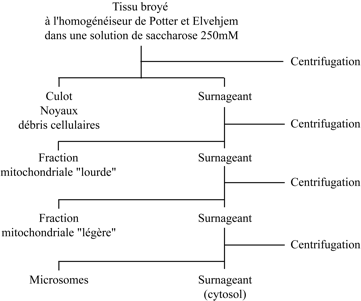 |
| Fractionnement subcellulaire d’un homogénat de foie par centrifugation différentielle. A chaque étape, la force centrifuge appliquée (vitesse et temps de centrifugation) augmente. Par rapport à la méthode originale de Claude (voir chapitre VI, paragraphe « Microsomes »), dans ce protocole, modifié par de Duve et Berthet, la fraction des gros granules est séparée en fraction mitochondriale lourde et fraction mitochondriale légère. |
Voir les lysosomes
Dans un article publié en 1953 dans The Journal of Histochemistry and Cytochemistry, Alex B. Novikoff (Albert Einstein College of Medicine) décrivait les résultats d’expériences de fractionnement subcellulaire : dans les cinq fractions particulaires isolées : nucléaire, mitochondriale, mixte, microsomiale I, microsomiale II, et dans le surnageant, il a dosé une série de constituants chimiques et enzymatiques choisi avec une remarquable pertinence car ils seront plus tard universellement utilisés comme « marqueurs » des principaux organites : ADN (noyaux), ARN (noyaux, reticulum endoplasmique rugueux, surnageant), succinate oxydase (mitochondries), phosphatase acide (lysosomes), uricase (peroxysomes), estérase (reticulum endoplasmique), phosphatase alcaline et 5’-nucléotidase (membrane plasmique). Les distributions dans les fractions se recouvraient largement les unes les autres et Novikoff en tira la conclusion que la composition biochimique des organites était hétérogène.
 |
| Lysosomes dans le cytoplasme d’un hépatocyte. Ils sont souvent localisés à proximité d’un canalicule biliaire. L’hétérogénéité du contenu est mise en évidence par une réaction histochimique en présence d’un sel de plomb. rer : reticulum endoplasmique rugueux. |
Rétrospectivement, il peut paraître surprenant que tant de biochimistes aient opté pour le postulat d’hétérogénéité ; ce postulat peut s’interpréter de deux façons : (i) les organites sont biochimiquement hétérogènes ; il y a « de tout partout » ; (ii) la composition d’un type d’organite (les mitochondries, par exemple) est hétérogène ; il y a des mitochondries avec beaucoup de succinate oxydase et d’autres qui en contiennent peu. La première hypothèse semble peu logique. Elle reviendrait à dire qu’un organite donné n’exerce pas de fonction spécifique dans la cellule, qu’il n’y a pas de relation entre structure et fonction.
Alex Novikoff comprit la portée de la découverte du Groupe de Louvain et accéda à la demande de Christian de Duve de collaborer à l’identification des lysosomes au microscope. Il profita de la tenue à Bruxelles du Third International Congress of Biochemistry, en 1955, pour faire un séjour dans le Laboratoire de Chimie physiologique, à Louvain. Henri Beaufay prépara des fractions enrichies en phosphatase acide (fraction L). Le laboratoire ne possédant pas de microscope électronique, Novikoff utilisa celui d’Albert Claude à l’Institut Bordet, à Bruxelles. Le contact entre les deux microscopistes ayant produit d’importantes décharges électriques, Novikoff effectua un repli stratégique vers l’Institut de Recherches sur le Cancer du Centre National de la Recherche Scientifique, à Villejuif, où il bénéficia de l’hospitalité de Wilhelm Bernhard. L’article publié en 1956 contenait une micrographie de la fraction L (light) montrant des vésicules au contenu hétérogène, de différentes tailles, entourées d’une membrane simple ; il contenait aussi une micrographie de Wilhelm Bernhard et Charles Rouiller montrant des vésicules semblables dans le cytoplasme d’hépatocyte de rat. Rouiller avait découvert ces organites en 1954 et, ignorant leur fonction dans la cellule, les avait baptisés « corps denses péri-canaliculaires » par référence à leur localisation préférentielle à proximité des canaux biliaires.
En 1961, Novikoff adapta à la microscopie électronique la technique de Gyorgy Gömöri (University of Chicago) pour révéler la présence de phosphatases dans les tissus. Le principe en était simple : l’hydrolyse du β-glycérophosphate libère du phosphate qui est capturé sous forme de sel de plomb, opaque aux électrons ; la transformation du phosphate de plomb en sulfure de plomb confère aux lysosomes une coloration brun foncé. La cellule renferme de nombreuses phosphatases ; pour éviter leur interférence dans la réaction cytochimique, le pH du milieu d’incubation est ajusté au pH optimum d’activité de la phosphatase acide (4,5). Les lysosomes purent ainsi être identifiés dans toutes les cellules eucaryotes (sauf les globules rouges), parfois jusqu’à plusieurs centaines par cellule. Ils présentent une grande hétérogénéité de taille, de forme et de contenu et sont entourés d’une membrane. A cause de leur caractère hétérogène (polydisperse, en jargon de physico-chimiste) il est difficile d’isoler par centrifugation une fraction de lysosomes dépourvue de contaminants, d’autant que les lysosomes sont des composants « mineurs » de la cellule (environ 1% des protéines cellulaires).
Références : Novikoff AB Biochemical heterogeneity of the cytoplasmic particles isolated from rat liver homogenates (1953)
Alex B. Novikoff AB, Henri Beaufay H, Christian de Duve C, Electron Microscopy of Lysosome-rich Fractions from Rat Liver, Journal of Biophysical and Biochemical Cytology (1956)
Gomori G Microtechnical demonstration of phosphatase in tissue sections (1939)
Gomori G Alkaline phosphatase of cell nuclei (1951)
L’appareil cellulaire de dégradation des macromolécules
Les lysosomes sont au cœur du système digestif de toutes les cellules, à l’exception des globules rouges ; ils dégradent les acides nucléiques, les protéines, les polysaccharides, les phospholipides, les sphingolipides, les glycoconjugués, internes ou importées, en libérant des micromolécules qui sont réutilisées par la cellule. Ils exercent leur fonction en milieu acide (qui dénature les protéines et les complexes protéiques et facilite ainsi leur digestion) ; ils constituent le principal compartiment acide de la cellule. Le pH à l’intérieur de ces organites est mesuré en introduisant une sonde fluorescente sensible au pH (Isothiocyanate de Fluorescéine couplé au dextran pour les lysosomes) ; on utilise aussi des sondes enrobées dans des liposomes et on mesure l’intensité de fluorescence par spectroscopie. Les résultats sont influencés par la concentration de la sonde fluorescente que l’on est parvenu à introduire dans l’organite. C’est ainsi que pour les lysosomes, les valeurs publiées dans la littérature varient de 3,5 à 5,0 et pour les endosomes, de 4,6 à 6,2. Pour s’affranchir de ce biais dans les résultats, on utilise des sondes ratio-métriques et on mesure un rapport d’intensités indépendant de la concentration. Pour les membranes cellulaires ou les organites entourés de membranes on dispose de sondes hydrophobes à base de 3-hydroxyflavone qui, après tautomérisation par transfert de proton, présentent deux bandes d’émission.
On a pu mettre en évidence l’existence d’une voie endocytaire « acide » qui prend naissance avec les vésicules d’endocytose, à partir d’invaginations de la membrane plasmique, passe par les endosomes précoces (pH +/- 6,5), les endosomes tardifs (pH +/- 5,5) et aboutit aux lysosomes (pH +/- 3,5). Ce processus graduel d’acidification facilite la dénaturation et la dégradation par les hydrolases acides des ligands internalisés par la cellule. Une ATPase membranaire transférant des protons du cytosol vers le milieu lysosomial et un canal à ions chlorure (Cl–) maintiennent l’acidité du contenu de l’organite. La fonction digestive est assurée par une impressionnante panoplie d’hydrolases : glycosidases (40), protéases (18), sulfatases (7), nucléases, lipases, phospholipases, phosphatases, saposines (Sphingo Activator PrO(S)teIN). Les lysosomes renferment aussi des protéines de structure (Lamp 1), de transport (Lamp 2), des canaux ioniques à chlorure et à calcium, des complexes cataboliques permettant la fusion de compartiment à compartiment.
Les lysosomes sont alimentés en matériaux à digérer venant de l’extérieur par les endosomes. Les macromolécules sont internalisées dans les cellules par endocytose « médiée » par récepteur. C’est le cas pour les lipoprotéines de faible densité circulant dans le sang, comme le montrèrent Richard G. Anderson, Michael S. Brown et Joseph L. Goldstein (University of Texas) ; ces deux derniers se virent attribuer le prix Nobel de médecine ou physiologie 1985. Les lipoprotéines, chargées de cholestérol, sont reconnues par un récepteur spécifique de la surface cellulaire. Un mécanisme similaire a été décrit pour la transferrine, une β-globuline qui transporte deux atomes de fer par molécule, de l’intestin vers le foie ou les réticulocytes. L’internalisation de la globuline et des atomes de fer complexés se fait par l’intermédiaire du récepteur de la transferrine inséré dans la membrane plasmique. Gilbert Ashwell et Anatol G. Morell (National Institute of Arthritis, Metabolism, and Digestive Diseases) montrèrent en 1968 que c’est également le cas pour la céruléoplasmine, une glycoprotéine de la circulation sanguine. La perte de l’acide sialique périphérique entraîne l’endocytose de la céruléoplasmine par un récepteur membranaire mis en évidence par Ashwell, et qui reconnaît le sucre périphérique des glycoprotéines circulantes et déclenche leur capture par la cellule (revue citée en référence).
Références : Brown MS, Goldstein JL Familial hypercholesterolemia: Defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity (1974)
Brown MS, Goldstein JL Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein (1976)
Anderson RG, Goldstein JL, Brown MS A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts (1977)
Ashwell G, Morell AG The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins (1974)
L’internalisation des macromolécules reconnues par les récepteurs de la membrane péricellulaire commence avec l’invagination de celle-ci au niveau des puits tapissés de molécules de clathrine. L’invagination s’accentue jusqu’à la formation d’une vésicule tapissée de clathrine formant un squelette (une « cage »). Les puits tapissés de clathrine furent observés au microscope électronique par Thomas Roth et Keith Porter en 1964 et isolés par Barbara M.F. Pearce (Laboratory of Molecular Biology, Medical Research Council, Cambridge) en 1975 à partir de tissus nerveux où ces organites sont abondants. En 1976, elle en purifia le principal constituant protéique, la clathrine, une protéine formée de trois branches (triskélion) ayant la propriété de s’assembler en une structure en forme de polyèdre. Le matériel endocyté dans les vésicules est dirigé vers les endosomes primaires où ils sont rejoints par les hydrolases acides synthétisées dans le reticulum endoplasmique et ayant transité par le trans-Golgi. Les endosomes primaires deviennent des endosomes secondaires ou des lysosomes. Les liquides, contenant généralement des substances dissoutes ou des substances adsorbées sur la membrane plasmique, sont intériorisés dans la cellule par un mécanisme particulier d’endocytose non spécifique appelé pinocytose.
Références : Roth TF, Porter KR Yolk protein uptake in the oocyte of the mosquito Aedes aegypti (1964)
Pearse BMF Clathrin: a unique protein associated with intracellular transfer of membrane by coated vesicles (1976)
Chez certaines cellules, les matériaux soumis à la digestion lysosomiale sont fournis par la phagocytose, une forme d’hétérophagie active. Elle fut observée pour la première fois en 1882 chez les larves mobiles d’étoile de mer par le zoologiste Ilya Ilyich Metchnikov (prix Nobel de médecine ou physiologie en 1908). Le sang circulant renferme des phagocytes « professionnels », les macrophages et les leucocytes neutrophiles, aussi appelés « polymorphonucléaires » par référence à la forme multilobée de leur noyau. Ces cellules jouent un rôle actif dans la défense de l’organisme, internalisant des bactéries ou des particules dans une structure entourée d’une membrane : le phagosome. Les lysosomes fusionnent avec le phagosome dont le contenu est digéré par les hydrolases. La phagocytose est très active chez les amibes, où elle joue le rôle de mécanisme nutritionnel plutôt que de mécanisme de défense.
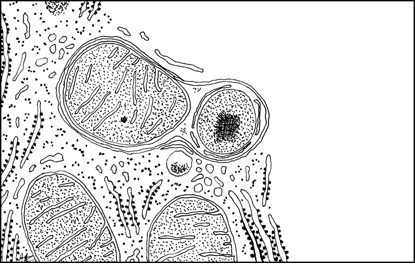 |
| Autophagosome renfermant une mitochondrie et un peroxysome. Les mitochondries ont une durée de vie qui est d’une dizaine de jours, en moyenne. |
Les lysosomes sont alimentés en peptides par les protéasomes, des complexes multienzymatiques présents dans le cytoplasme et le nucléoplasme. Ces complexes dégradent les protéines mal formées, mal repliées ou arrivées en fin de cycle ; les peptides libérés sont porteurs d’une étiquette (une séquence d’acides aminés) qui permet leur reconnaissance par des perméases de la membrane lysosomiale. Dans les lysosomes, ils sont hydrolysés par les peptidases en acides aminés. Ceux-ci, comme les autres produits d’hydrolyse : acides gras, sucres, nucléotides, passent dans le cytosol en franchissant la membrane des lysosomes par diffusion ou transport assisté. Ils sont réutilisés pour synthétiser des constituants cellulaires, ou dégradés avec récupération d’énergie.
Les lysosomes sont aussi alimentés en substrats à dégrader par les autophagosomes. L’autophagie est un mécanisme d’autodestruction contrôlée de portions de la cellule. Ce mécanisme fut découvert en 1962 par Thomas P. Ashford, un étudiant du laboratoire de Keith Porter (The Rockefeller Institute for Medical Research), dans les hépatocytes d’animaux ayant reçu une injection de glucagon. Une membrane issue du reticulum endoplasmique séquestre une partie du cytoplasme donnant ainsi naissance à une vacuole de grande taille renfermant des organites. Porter pensait être en présence du mécanisme de formation des lysosomes. En 1963, Christian de Duve inventa le terme « autophagie » pour décrire la fusion des autophagosomes avec des lysosomes et la dégradation du contenu de la vacuole par les hydrolases lysosomiales. La macro-autophagie joue un rôle dans la destruction d’organites endommagés ou en fin de vie, l’autodestruction contrôlée de certains tissus et l’involution de certains organes comme le rein. Au cours de l’embryogenèse, le tissu rénal est partiellement détruit puis reconstruit pour former le rein mature. La macro-autophagie intervient dans le renouvellement des organites subcellulaires : dans des conditions de nutrition normales, elle assure le renouvellement, chaque semaine, de la majeure partie des composants d’un hépatocyte. La macro-autophagie apparaît ainsi comme le pivot de l’homéostasie cellulaire. C’est aussi un mécanisme de survie des cellules en souffrance, par exemple par dénutrition après un jeune prolongé. Les cellules consomment alors leurs propres constituants. Enfin, c’est un mécanisme de défense de la cellule contre les infections et l’invasion par des bactéries.
Référence : Ashford TP, Porter KR Cytoplasmic Components in Hepatic Cell Lysosomes (1962)
A côté de la voie principale de la macro-autophagie, on distingue une micro-autophagie et une autophagie assistée par des chaperones. Dans la micro-autophagie, il n’y a pas de formation de vésicules préalablement à la fusion avec les lysosomes ; le matériel cytoplasmique pénètre directement par invagination de la membrane lysosomiale. A la différence des macro- et micro-autophagies, l’autophagie médiée par une chaperone est un phénomène sélectif. Le matériel à ingurgiter passe les complexes hsc70 (heath shock cognate) du cytoplasme et des lysosomes. Quelle que soit sa forme, l’autophagie, comme le fonctionnement du système lysosomial dans son ensemble, sont des phénomènes étroitement régulés selon les tissus, l’âge des cellules et l’environnement. Au début des années 1980, à l’Institut de Technologie de Tokyo (Tokyo Tech), Yoshinori Ohsumi (prix Nobel de physiologie ou médecine en 2016) étudia l’autophagie chez la levure Saccharomyces cerevisiae soumise au jeune. En 1992, il publia ses résultats sur l’identification des 15 gènes impliqués dans le processus.
A la fin de ce sous-chapitre, je voudrais faire mention du rôle joué par les lysosomes dans la production de peptides antigéniques. Les lymphocytes T CD4 (lymphocytes T auxiliaires, helper) sont des acteurs majeurs de la réponse immunitaire. Ils reconnaissent les peptides antigéniques présentés par des molécules d’histocompatibilité de classe II – chez l’homme HLA-DR, DP, DQ – à la surface des cellules présentatrices d’antigènes (macrophages, cellules dendritiques, lymphocytes B activés). Les lymphocytes T CD4 agissent en activant d’autres cellules du système immunitaire : ils dirigent les lymphocytes B dans leur différenciation vers la production d’anticorps.
Les molécules du complexe majeur d’histocompatibilité II sont des glycoprotéines dimériques, dotées d’un peptide signal amino-terminal ; elles sont synthétisés dans le domaine ribosomal du reticulum endoplasmique, où elles s’associent à une protéine membranaire de type 2, la chaîne invariante ; la fonction de cette chaperonne est double : (i) protéger les sites de fixation des peptides contre toute association prématurée avec des peptides (ii) accompagner le complexe au cours des étapes de glycosylation dans le reticulum endoplasmique et le Golgi. Doté d’un second signal topogénique, le signal mannose 6-phosphate, le complexe atteint le réseau trans-golgien, d’où il est dirigé vers les endosomes. A pH acide, les protéases endosomales dégradent la chaîne invariante laissant un peptide (class II associated invariant chain peptide, CLIP) associé à la fente des chaînes α et β. Les peptides reconnus par les lymphocytes T auxiliaires sont présents dans le compartiment endosomes – lysosomes. Ils ont été générés à partir d’antigènes endocytés – ou phagocytés – par les cellules présentatrices et dégradés par les protéases à pH acide. C’est dans ce compartiment que les molécules du complexe majeur d’histocompatibilité II rencontrent les peptides antigéniques et que se fait l’échange CIP – peptide.
Note : Pour la rédaction de ce paragraphe, les informations m’ont été fournies par le Dr. Pierre Coulie (de Duve Institute, Bruxelles), qui en a aussi supervisé le texte.
Fonctions des Lysosomes
| Renouvellement (turn over) des constituants et des protéines intra- et extra-cellulaires |
|---|
| Dégradation et recyclage des organites cellulaires |
| Maintien de l’homéostasie cellulaire |
| Contrôle de qualité des protéines |
| Dégradation des protéines mal formées ou anormales |
| Dégradation des récepteurs non recyclés de la membrane plasmique |
| Réparation des membranes cellulaires |
| Fusion des lysosomes avec la membrane endommagée |
| Invagination de la membrane et endocytose du site de la lésion |
| Homéostasie du cholestérol et maintien de l’intégrité de la membrane plasmique |
| Endocytose des lipoprotéines (LDL) plasmatiques |
| Transfert vers la membrane plasmique du cholestérol libéré dans les lysosomes |
| Remodelage du squelette osseux |
| Hydrolyse de protéines de la matrice extracellulaire osseuse par les ostéoclastes |
| (Cathepsine K, phosphatase acide pourpre) |
| Maturation des pro-hormones |
| (production de thyroxine dans la lumière des follicules thyroïdiens par protéolyse extracellulaire de la thyroglobuline par la cathepsine K) |
| Digestion des bactéries et autres agents pathogènes par les hydrolases lysosomiales dans les phagosomes des macrophages |
| Production de peptides antigéniques reconnus par des lymphocytes T |
| Voie de présentation par les molécules du Complexe majeur d’histocompatibilité de classe II |
| Régulation de la sécrétion des cellules hématopoïétiques |
| (stockage dans les lysosomes des produits de sécrétion) |
Clairance cellulaire
Les travaux d’Andrea Ballabio et de ses associés (Thelethon Institute of Genetic and Medicine, Università di Napoli Federico II) ont jeté un éclairage nouveau sur le rôle central joué par les lysosomes dans la clairance cellulaire. La protéine Lamp 1 (Lysosomal Assiociated Membrane Protein 1) est une glycoprotéine membranaire utilisée au laboratoire comme marqueur de différentiation des cellules tumorales. En 2009, Carmine Settembre, Alessandro Fraldi et Diego L. Medina découvrirent dans le promoteur de Lamp 1 la présence de séquences E-box (Enhancer box) ; ces séquences palindromiques sont des sites de fixation sur l’ADN d’un facteur de transcription régulant l’expression des gènes :
![]()
N désigne indifféremment l’un des quatre nucléotides. Le facteur de transcription a été identifié : c’est un hétéro-dimère de la sous famille des facteurs de transcription à motif hélice – boucle – hélice et tirette à leucines (leucine zipper). Baptisé TFEB, le facteur est localisé dans la membrane des lysosomes ; il déclenche la transcription des gènes lysosomiaux en se fixant sur les sites E-box de leur promoteur. Dans les cellules où TFEB est surexprimé, on observe une augmentation du nombre de lysosomes et une activité dégradative accrue (régulation positive). L’ensemble forme un réseau de gènes (CLEAR, Coordinated Lysosomal Enhancement And Regulation) impliqués dans la biogenése et l’activité des lysosomes.
En 2011, C. Settembre et collaborateurs montrèrent que le facteur de transcription EB contrôle aussi l’autophagie. En présence de molécules à dégrader, une sérine/thréonine kinase membranaire (mTOR, Mammalian Target Of Rapamycin) inactive TFEB en le phosphorylant sur une sérine ; en l’absence de molécules à digérer (jeune), TFEB est activé par une phosphatase (la calcitonine activée par les ions Ca++) et franchit l’enveloppe nucléaire pour déclencher la transcription des gènes lysosomiaux, l’autophagie et la coopération entre lysosomes et phagosomes pour dégrader les macromolécules endogènes. L’état du stock de nutriments cellulaires est apprécié par une machinerie protéique membranaire appelée LYNUS (Lysosome NUtrient Sensing machinerie), à laquelle appartiennent mTOR, et les ATPases v-ATPase et Na ATPase lysosomiale.
Les lysosomes de certaines cellules pratiquent une forme de digestion extracellulaire. Les ostéoclastes et les chondroclastes sont des macrophages qui participent au remodelage et au renouvellement du tissu osseux en exportant dans le milieu extracellulaire des hydrolases lysosomiales et un suc acide qui dégradent les matrices osseuse et cartilagineuse. Le suc acide dissout l’hydroxyapatite, une combinaison de phosphate et d’hydroxyde de calcium, qui constitue la partie minérale de l’os. L’échafaudage de collagène est attaqué par les enzymes. Les fragments sont intériorisés dans les ostéoclastes et digérés dans les lysosomes. Les matrices osseuse et cartilagineuse sont ensuite reconstruites par les ostéoblastes et les chondroblastes. Dans la thyroïde, les lysosomes interviennent dans la sécrétion hormonale. La thyroglobuline synthétisée dans les cellules de l’épithélium thyroïdien est sécrétée dans les follicules thyroïdiens où elle subit une iodination. Elle est intériorisée par endocytose dans les cellules épithéliales. Les endosomes fusionnent avec les lysosomes où la thyroglobuline est dégradée avec libération de thyroxine. Les cellules épithéliales libèrent l’hormone thyroïdienne dans la circulation sanguine.
Pathologie des lysosomes (Lysosomal storage disorders)
Maladies neurodégénératives
La première maladie liée à un dysfonctionnement des lysosomes fut découverte au milieu des années 1950. La glycogénose de type II est une affection grave qui se traduit par une accumulation de glycogène dans le cœur, les muscles et des organes comme le foie. Les premiers cas cliniques furent décrits en 1933, aux Pays-Bas, par le pédiatre Johan C. Pompe (la glycogénose de type II est appelée maladie de Pompe) et en Allemagne par G. Bishof et W. Putschar. La biochimiste Gerty Th. Cori (Washington University, Saint Louis) identifia à partir d’échantillons provenant de trois glycogénoses l’enzyme dont l’absence ou la déficience est responsable de l’accumulation de glycogène dans les tissus. Cet enzyme appartient à la voie de dégradation du glycogène. Henry Géry Hers (Laboratoire de Chimie physiologique, Université catholique de Louvain) montra, en 1954, que dans de la maladie de Pompe la déficience porte sur un enzyme qui, dans les conditions habituelles, joue un rôle tout à fait secondaire dans le catabolisme du glycogène : l’α-1,4-glucosidase hydrolyse à pH 5 les disaccharides comme le maltose (d’où son nom de « maltase acide »), les oligosaccharides et les polysaccharides. Le fait que cette hydrolase ait un pic d’activité à pH acide attira l’attention de Christian de Duve : le fractionnement par centrifugation d’homogénat de foie montra une distribution superposable à celle de la phosphatase acide. L’examen au microscope électronique de biopsies provenant d’un jeune patient atteint de la maladie de Pompe montra la présence dans les cellules hépatiques de lysosomes gorgés de rosettes de glycogène. La présence d’α-1,4-glucosidase dans les lysosomes révéla l’existence de deux voies métaboliques de dégradation du glycogène en glucose : une voie « normale » passant par la glycogène phosphorylase cytosolique, et une voie passant par l’autophagie. Lorsque le glycogène s’accumule dans le cytosol, il est séquestré dans des vacuoles et dégradé par la maltase acide des lysosomes.
La glycogénose de type II s’inscrit dans le cadre général des erreurs innées du métabolisme (Inborn error of metabolism) découvertes par Archibald Garrod. Ce sont des maladies de surcharge des lysosomes dans lesquelles une déficience génétique a entraîné l’absence d’une hydrolase acide, ce qui provoque l’accumulation de déchets non digérés. Les conséquences du dysfonctionnement des lysosomes entraînent le dysfonctionnement d’un ou de plusieurs organes. On a identifié plus de 50 thésaurismoses dont la gravité témoigne s’il en était besoin de l’importance du rôle des lysosomes dans la clairance cellulaire et l’élimination des déchets. Ce sont des maladies héréditaires récessives parmi lesquelles on a dénombré une quinzaine de lipidoses caractérisées par de graves dysfonctionnements du système nerveux : maladie de Gaucher (déficit en glucocérébrosidase), maladie de Tay-Sachs (déficit en héxosaminidase A), maladie de Niemann-Pick (déficit en sphingomyélinase) etc. Les stratégies de correction de ces déficits en enzymes font appel à la transplantation médullaire ou à la thérapie enzymatique substitutive. Elles ont été essayées dans la maladie de San Filippo A (déficit en héparane sulfamidase) et dans la leucodystrophie métachromatique (déficit en aryl sulfatasse A). Un grand espoir dans la correction de ces pathologies est suscité par la stratégie mise en œuvre dans le groupe d’Andrea Ballabio pour stimuler la clairance des déchets accumulés.
Les maladies neuro-dégénératives avec accumulation de dépôts intracellulaires d’agrégats de protéine bêta-amyloïde et de protéine tau (maladie d’Alzheimer) ou de protéines anormales (maladies de Parkinson ou de Huntington) pourraient bénéficier de la même approche thérapeutique.
Thérapie lysosomotrope
Les protozoaires flagellés Leishmania transmettent à l’homme des infections graves, voire mortelles. Leishmania donovani infecte les macrophages du foie, de la rate et de la moelle osseuse en passant par le système phagosomes – lysosomes. D’autres protozoaires, responsables d’affections parasitaires, passent aussi par le système vacuolaire et par les phagosomes – lysosomes : Toxoplasma gondii (toxoplasmose), coccidies (coccidiose), Trypanosoma cruzi (maladie de Chagas), Plasmodium (malaria). Une thérapie lysosomotrope fut envisagée pour combattre ces parasitoses ; elle consiste à faire pénétrer par endocytose dans les cellules infectées des drogues anti-parasitaires couplées à des vecteurs macromoléculaires,; après hydrolyse du vecteur, ou de la liaison vecteur-drogue par les enzymes lysosomiaux, la substance anti-parasitaire est libérée dans un espace confiné, en contact avec le parasite.
Références : De Duve C, Trouet A Lysosomes and lysosomotropic drugs in host-parasitic relationship (1973)
Jadin JM, Trouet A, Van Hoof F, Bioul-Marchand M, Maldague P, Jadin-Nyssens M Étude comparative d’une chimiothérapie lysosomotrope dans la maladie de Chagas et dans le Nagana (1977)
Partant du même principe, André Trouet et Christian de Duve (Laboratoire de Chimie physiologique, Université Catholique de Louvain) imaginèrent une thérapie lysosomotrope pour traiter certaines formes de cancers. De nombreuses substances antinéoplasiques utilisées en thérapie anti-tumorale sont aussi des cytostatiques ; elles bloquent l’activité proliférative des cellules cancéreuses, c’est-à-dire leur capacité à se diviser. Les antinéoplasiques manquent de spécificité ; les cellules germinales, les cellules souches des globules blancs et des globules rouges et d’une manière plus générale les cellules des tissus à taux de renouvellement élevé – sang, peau, muqueuses -, sont aussi des cibles. De plus, pour un certain nombre d’agents anti-tumoraux, la différence entre dose thérapeutique et dose toxique est faible. Dans les années 1960-1970, on était en quête d’une thérapie anti-tumorale efficace et spécifique, ciblant les cellules tumorales à l’exclusion des cellules saines. A côté de la recherche de nouveaux agents par extraction et criblage à partir de matériels divers – une approche longue et coûteuse -, et de perspectives thérapeutiques nouvelles résultant d’une meilleure connaissance des mécanismes moléculaires et cellulaires de la cancérisation, s’ouvrait l’option de modifier la structure des antinéoplasiques existants pour les rendre moins toxiques et plus efficaces, par exemple par couplage à des macromolécules.
Références : de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, Van Hoof F Lysosomotropic agents (1974)
Trouet A, Deprez-de Campeneere D, Maldague P, Jadin JM, Van Hoof F The Concept of Lysosomotropic Chemotherapy: Applications to Neoplastic and Parasitic Diseases (1977)
Trouet A Carriers for drugs (1977)
La « thérapie lysosomotrope » imaginée par Trouet et de Duve est essentiellement basée sur l’idée que l’endocytose est plus élevée chez les cellules tumorales que chez les cellules dites normales. Vers le milieu des années 1950, la croyance s’est imposée que les besoins en protéines des cellules tumorales dépassent leur capacité à les synthétiser ; pour compenser le déficit, les cellules tumorales à croissance rapide sont des « pièges à azote » qui captent, par pinocytose, des protéines dans le milieu extracellulaire et le plasma. Un certain nombre de résultats font état d’un taux de captation des protéines par pinocytose élevé dans les cellules de tumeurs expérimentales ou humaines : leucémie, lymphome, réticulosarcome, hépatome, glioblastome ; je cite quelques articles sur le sujet :
Références : Cohen S, Beiser SM, Hsu KC Immunohistochemical study of the uptake of serum proteins by neoplastic liver cells (1961)
Ghose T, Nairn RC, Fothergill JE Uptake of proteins by malignant cells (1962)
Easty GC, Yarnell MM, Andrews RD The uptake of proteins by normal and tumour cells in vitro (1964)
Ioverno L, Levis A, Palumbo A, Ghezzo F, Genetta C, Pegoraro L Phagocytic activity and cytochemical characterization of acute human myeloblastic leukemia cells (1982)
Voir aussi: Anderson D, Bush H, Greene HS, Simbonis S Studies on the metabolism of plasma proteins in tumor-bearing rats. II. Labeling of intracellular particulates of tissues by radioactive albumin and globulin (1956)
Henderson JF, LePage GA The Nutrition of Tumors: A Review (1959)
Lepage GA, Henderson JF Biochemistry of tumors (1960)
Busch H, Fujiwara E, Firszt DC Studies on the metabolism of radioactive albumin in tumor-bearing rats (1961)
En théorie, le ciblage d’une substance chimiothérapique nécessite qu’elle soit liée à un ligand reconnu par un récepteur spécifique des cellules tumorales. La mise au point pratique d’une telle stratégie représente un travail de très longue haleine. L’approche choisie par Trouet, basée sur la différence d’activité endocytaire entre cellules normales et cancéreuses, vise à court-circuiter ce laborieux processus. Elle est plus rapide mais présente le défaut d’être quantitative et non qualitative comme dans le modèle ligand-récepteur spécifique. Le vecteur macromoléculaire sélectionné fut l’ADN et l’antinéoplasique, les anthracyclines daunomycine (daunorubicine) ou adriamycine (doxorubicine, un inhibiteur de la topo-isomérase II). La daunomycine pénètre dans les cellules par diffusion. Son emploi est limité par sa toxicité : injectée par voie intramusculaire elle provoque des nécroses musculaires, notamment du muscle cardiaque. La formation du complexe ADN-daunorubicine se fait par contact entre les deux ingrédients dans des conditions définies ; la daunorubicine s’intercale dans le petit sillon de l’ADN. Un certain nombre de conditions doivent être remplies : (i) le complexe doit être stable dans le sang et les milieux intracellulaires ; (ii) le complexe doit pénétrer dans les cellules par endocytose ou pinocytose ; on a pu le montrer sur des cellules L1210 en culture ; (iii) après passage des endosomes aux lysosomes, l’association vecteur – agent anti-tumoral doit être dissociable, libérant ainsi le principe actif ; le vecteur ADN est digéré par les DNAses lysosomiales ; (iv) après hydrolyse du vecteur, l’agent thérapeutique libéré doit être résistant à la digestion par le vaste éventail des enzymes hydrolytiques des lysosomes.
Dans l’espace réduit des lysosomes, les anthracyclines atteignent des concentrations élevées et fragilisent la membrane lysosomiale,ce qui entraîne une libération de cathepsines dans le cytoplasme. On contourne ainsi un obstacle majeur auquel se heurte la chimiothérapie classique : en activant la mort cellulaire par rupture des lysosomes, on compense le blocage de la voie classique de l’apoptose chez les cellules tumorales. Des essais thérapeutiques furent entrepris, d’abord sur des cellules L1210 en culture, puis sur la forme intrapéritonéale de la leucémie L1210 chez la souris DBA/2 (Dilute Brown Non-Agouti) et enfin chez des patients. Après des débuts encourageants et quelques succès initiaux, les essais en clinique humaine durent être abandonnés. La liaison ADN-daunorubycine n’est pas aussi stable dans le plasma sanguin qu’on le croyait, ce qui explique peut-être pourquoi des chercheurs du National Cancer Institute (Bethesda), intéressés par le ciblage (targeting) de substances anti-tumorales, ne sont pas parvenus pas à reproduire les résultats de Trouet et de Duve. D’autres part, l’injection d’ADN a des patients n’est pas sans soulever de multiples problèmes, notamment sur le plan éthique. C’est pourquoi la daunorubicine a été ultérieurement conjuguée à la sérum albumine par une liaison amide entre le groupe aminé de l’anthracycline et un groupe carboxyle de la protéine. L’intercalation d’un espaceur de trois acides aminés entre la protéine et la drogue augmente la sensibilité à l’hydrolyse dans les lysosomes.
Références : Deprez-de Campeneere D, Trouet A DNA-anthracycline complexes. I. Toxicity in mice and chemotherapeutic activity against L1210 leukemia of daunorubicin-DNA and Adriamycin-DNA (1965)
Trouet A, Deprez-De Campeneere D, de Duve C Chemotherapy through Lysosomes with a DNA-Daunorubycin Complex (1972)
Langslet A, Oye I, Lie SO Decreased cardiac toxicity of Adriamycin and daunorubicin when bound to DNA (1974)
Cornu G, Michaux JL, Sokal G, Trouet A Daunorubicin-DNA : Further Clinical Trials in acute non-lymphoblastic leukemia (1974)
Trouet A, de Duve C, De Smet-Malengreaux M, Atassi G Experimental leukemia chemotherapy with a « lysosomotropic » adriamycin-DNA complex (1974)
Trouet A, Sokal G Clinical Studies with Daunorubicin-DNA Complexes: A Review (1974)
Longueville J, Maisin H Combined Therapy with Adriamycin-DNA, Vincristine and Medoxy-Progesterone Acetate in Metastatic Breast Cancer (1975)
Michaux JL, Cornu G, Sokal G, Trouet A Preliminary clinical trials with the adriamycin-DNA complex in human leukemias and non-Hodgkin lymphomas (1975)
Deprez-De Campeneere D, Baurain R, Huybrechts M, Trouet A A Comparative study in mice of the toxicity, pharmacology, and therapeutic activity of daunorubicin-DNA and doxorubicin-DNA complexes (1979)
Trouet A, Masquelier M, Baurain R, Deprez-De Campeneere D A covalent linkage between daunorubicin and proteins that is stable in serum and reversible by lysosomal hydrolases, as required for a lysosomotropic drug-carrier conjugate: in vitro and in vivo studies (1982)
Note. L’implication des lysosomes dans une thérapie anti-tumorale continue à faire l’objet de recherches. Je cite comme exemple les travaux de Teodora Krol et de Wojciech et Ewa Trybus (Jan Kochanowski University, Kielce) :
Trybus W, Trybus E, Krol T Lysosomes as a Target of Anticancer Therapy (2023)