Peroxysomes
« Microbodies »
Dans sa thèse de doctorat présentée en 1954 au Karolinska Institutet, à Stockholm, Johannes A.G. Rhodin décrivit la présence dans les cellules des tubes contournés du rein de souris, d’organites de forme plus ou moins sphérique, avec une matrice granulaire entourée d’une membrane ; il les appela « microbodies ». Des corpuscules similaires furent observés dans les hépatocytes par Hédi Gänsler et Charles Rouiller (Institut de Recherches sur le cancer, Villejuif), dans les cellules végétales (cotylédons) par Hilton Mollenhauer (en 1966) et chez les protozoaires par Miklos Müller (en 1968). Dans le foie, mais pas dans le rein, ces organites ont une matrice contenant une structure para-cristalline d’aspect lamellaire : le nucléoïde. Wilhelm Bernhardt et Charles Rouiller notèrent une prolifération des microbodies dans le foie en cours de régénération ; ils en conclurent que ces organites étaient les précurseurs des mitochondries et que le nucléoïde était le précurseur des crêtes mitochondriales.
Références : Rhodin J. Correlation of ultrastructural organization and function in normal and experimentally changed proximal convoluted tubule cells of the mouse kidney (1954)
Rouiller C, Bernhard W. “Microbodies” and the problem of mitochondrial regeneration in liver cells (1956)
Granules à uricase
Dans le courant des années 1960, des études morphologiques, biochimiques et histochimiques établirent l’existence de granules à uricase dans un certain nombre de cellules. Zdenek Hruban et Hewson Swift (Department of Pathology, University of Chicago) montrèrent que la structure paracristalline (nucléoïde) de ces granules est constituée d’uricase. L’urate oxydase est le premier enzyme d’une courte voie catabolique allant de l’acide urique à l’allantoïne ; cet enzyme à cuivre catalyse l’oxydation de l’urate selon la réaction :
![]()
L’urate oxydase est présente chez les êtres vivants, des bactéries aux mammifères. Chez ceux qui en sont dépourvus (homme, grands singes, oiseaux, reptiles), les peroxysomes n’ont pas de nucléoïde et le produit final du catabolisme des purines n’est pas l’allantoïne mais l’acide urique.
Alex B. Novikoff, Estelle Podber et Jean Ryan (University of Vermont College of Medicine) fractionnèrent par centrifugation des homogénats de foie de rat et montrèrent que l’uricase est liée à une structure subcellulaire dont la distribution dans les fractions est intermédiaire entre celle de la cytochrome oxydase (mitochondries) et celle de l’estérase (microsomes). A. L. Tappel publia en 1963 une méthode de purification par centrifugation de lysosomes de foie de rat débarrassés de mitochondries et de granules à uricase (« Uricase Particles »). Dans les hépatocytes, ces organites ont à peu près la même taille que les lysosomes : environ 0,5 micromètre (de 0,1 à 1 micromètre) et un coefficient de sédimentation du même ordre de grandeur.
Références : Hruban Z, Swift H Uricase : localization in hepatic microbodies (1964)
Novikoff AB, Podber E, Ryan J Phosphatases of rat liver: II. Adenosintriphosphate dephosphorylation in regenerating liver (1953)
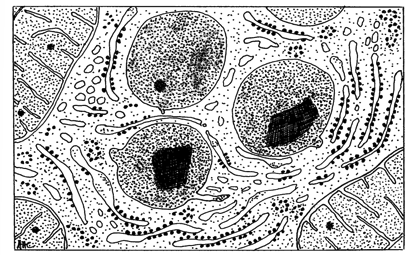 |
| Peroxysomes. La structure paracristalline présente dans la matrice est constituée d’urate oxydase. |
Après fractionnement d’un homogénat par centrifugation différentielle, cytochrome oxydase, phosphatase acide et uricase sédimentaient essentiellement dans la fraction mitochondriale. Mais si l’on séparait de la fraction mitochondriale une fraction lourde (M) et une fraction légère (L), ces trois enzymes se dissociaient partiellement les uns des autres, ce qui suggèrait l’existence de deux ou plusieurs entités différentes. Deux autres oxydases, la catalase et la D-amino acide oxydase, se comportaient comme l’uricase. La catalase, découverte au début du XXe siècle, a pour fonction de détruire l’eau oxygénée toxique pour les cellules qui se forme au cours de certaines réactions métaboliques. En fractionnement par centrifugation différentielle, la catalase se comportait à peu près comme les enzymes lysosomiaux ; elle présentait comme ces derniers le phénomène de latence[1] ce qui en faisait une candidate pour l’arsenal enzymatique des lysososomes, sauf que ce n’était pas une hydrolase.
La fraction mitochondriale renferme trois organites différents
En 1955, dans le processus de purification de la glucose 6-phosphatase microsomiale, Henri Beaufay (Laboratoire de Chimie physiologique, Université catholique de Louvain) utilisait un acide biliaire, le désoxycholate, pour la détacher de la membrane du reticulum endoplasmique. Le désoxycholate est doté d’un fort pouvoir détergent mais il est difficile à éliminer de la préparation purifiée, ce qui est un inconvénient majeur car il exerce un effet inhibiteur sur l’enzyme. Jacques Berthet suggéra de remplacer le désoxycholate par de la digitonine, une saponine de structure complexe extraite des graines de digitale ; mise en présence de matériel biologique, ce glucoside a deux effets : à faibles doses il forme un complexe cristallin avec le cholestérol pour lequel il présente une grande affinité ; à doses plus élevées il exerce un effet détergent, d’où son emploi pour solubiliser les protéines membranaires. La digitonine peut être éliminée en ajoutant du cyclohexanol au milieu : il se forme un complexe digitonine – cyclohexanol qui cristallise et précipite.
En 1956, au cours d’une étude de la latence des enzymes de la fraction mitochondriale, Beaufay délaissa les détergents utilisés habituellement (Triton X-100 ou désoxycholate) au profit de la digitonine. Avec la collaboration de Michel Caudron, un étudiant en médecine en stage au laboratoire, il traita une fraction mitochondriale avec des quantités croissantes de digitonine ; après centrifugation, il dosa dans le surnageant l’activité de trois enzymes : catalase, glutamate déshydrogénase (mitochondries) et phosphatase acide (lysosomes). La comparaison des courbes de solubilisation montrait que la phosphatase acide était libérée dans le surnageant à une concentration en saponine vingt fois plus faible que celle nécessaire pour libérer la catalase et la glutamate déshydrogénase. Le résultat de cette expérience montrait clairement que la phosphatase acide et la catalase (qui présentaient toutes deux le phénomène de latence) n’étaient pas localisées dans le même organite. C’était la première indication qu’il existait dans la fraction mitochondriale un troisième organite différent des mitochondries et des lysosomes. Cette expérience fut reproduite par Pierre Baudhuin dans le laboratoire de Christian de Duve à Rockefeller University et le résultat, publié en 1965 dans les Harvey Lectures sous le titre « The Separation and Characterization of Subcellular Particles ».
Centrifugation isopycnique en gradient de densité
Les résultats d’expériences de centrifugation différentielle montraient des différences de comportement entre la cytochrome oxydase, la phosphatase acide et l’uricase. Cependant les coefficients de sédimentation étaient trop proches les uns des autres pour permettre une distinction nette. Il fallait trouver une méthode de séparation basée sur un autre critère que le volume des particules. La centrifugation différentielle sépare les organites essentiellement en fonction de leur taille (plus précisément de leur volume) alors qu’en centrifugation en gradient de densité, le critère de séparation est la densité d’équilibre de la particule dans le milieu utilisé (on parle de centrifugation isopycnique). En 1957, Henri Beaufay entreprit une adaptation technique de la centrifugation en gradient de densité à la séparation des organites subcellulaires. Il fut rejoint par Jacques Berthet qui venait de terminer un séjour dans le laboratoire d’Earl Sutherland (Department of Pharmacology, Western Reserve University School of Medicine), où il avait étudié les effets métaboliques du glucagon.
A cette époque, la centrifugation en gradient de densité en était encore au stade artisanal. Elle commençait à être utilisée dans un certain nombre de laboratoires depuis la commercialisation, en 1953, de rotors à godets basculants. La méthodologie la plus utilisée pour préparer les gradients de densité (parce que la plus facile) consistait à superposer des couches de saccharose de densités croissantes et à attendre que la diffusion atténue les différences aux interfaces. Cette technique présentait un inconvénient majeur, à la source d’artefacts de séparation : les gradients obtenus n’étant pas linéaires, la répartition des organites ne coïncidait pas strictement avec leur distribution de densité. A chaque interface entre deux couches de saccharose de densités différentes, se produisait une accumulation d’organites formant une « couche ». C’est ainsi que pendant des années on a préparé – et on continue à le faire – des « microsomes lisses » et des « microsomes rugueux ». Le problème des séparations factices fut résolu par la construction d’une machine à cames délivrant un gradient linéaire en fonction du volume. Le recueil des fractions se faisait habituellement par aspiration du liquide à partir du ménisque du gradient ou par collecte des gouttes passant par un orifice percé à la base du tube. Ces procédés provoquaient des convections au sein du gradient et un mélange partiel des couches de densités différentes. Pour pallier à cet inconvénient, Beaufay construisit un sectionneur de tube. Christian de Duve et Jacques Berthet résumèrent les aspects théoriques et pratiques de la centrifugation en gradient de densité dans un volumineux article de revue : « Gradient centrifugation of cell particles. Theory and applications ».
La centrifugation en gradient de densité fut appliquée à l’analyse de la fraction mitochondriale. Les premiers résultats furent présentés au IVe Congrès international de biochimie, à Vienne en 1958. Les particules à uricase se dissociaient nettement des mitochondries et partiellement des lysosomes dans des gradients de saccharose dans l’eau lourde (à la différence de la membrane des lysosomes, celle des peroxysomes est perméable au saccharose, ce qui leur confère une densité d’équilibre élevée). Ces expériences furent répétées dans des conditions expérimentales où le choc osmotique subi par les granules dans un gradient de saccharose (un soluté d’osmolarité élevée) était minimisé. En effet, en migrant à travers le gradient des couches de faible densité vers les couches de densité élevée, la pression osmotique à laquelle les granules sont soumis était de plus en plus élevée, ce qui provoquait des artefacts de comportement. Pour palier à cet inconvénient, Beaufay utilisa comme soluté un composé macromoléculaire (le glycogène ne traverse pas les membranes) dissous dans une solution iso-osmotique de saccharose. Différentes expériences de centrifugation isopycnique furent conduites à différentes osmolarités. Les résultats, publiés[2] en 1964, apportèrent une seconde preuve de la coexistence dans les gros granules de trois populations distinctes d’organites : mitochondries, lysosomes et granules à uricase, catalase et D-aminoacide oxydase. Pierre Baudhuin ajoutera une quatrième oxydase : la lactate oxydase.
Triton WR-1339
Au début des années 1960, les recherches au Laboratoire de chimie physiologique portaient sur la stabilité de la membrane lysosomiale. Christian de Duve avait formulé l’hypothèse des « suicide bags » : la rupture accidentelle de la membrane lysosomiale provoquerait la libération dans le milieu intracellulaire d’hydrolases, exposant ainsi la cellule à un danger mortel. Nous savons aujourd’hui que cette crainte était largement injustifiée : les hydrolases sont actives à pH 3,5-5,0 (pH intra-lysosomial) ; elles le sont beaucoup moins au pH du cytosol : 7,2 à 7,4. Les lysosomes n’attaquent les structures cellulaires que dans les tissus nécrosés et mal vascularisés dans lesquels la diminution du pH intracellulaire altère la membrane lysosomiale et libère les hydrolases dans le cytoplasme. La dénomination « suicide bags » pour désigner les lysosomes fut abandonnée afin d’éviter toute confusion avec le suicide des cellules par apoptose, un processus physiologique de mort cellulaire génétiquement programmée.
En recherchant des substances affectant la stabilité de la membrane lysosomiale, Robert Wattiaux (Laboratoire de Chimie physiologique, Université catholique de Louvain) observa que l’addition de cholestérol à une fraction enrichie en lysosomes exerçait un effet stabilisateur, mesuré par une diminution de la latence ; par contre, un détergent non-ionique, le Triton WR1339, avait un effet déstabilisateur. Ce dérivé du polyéthylène glycol avait fait l’objet de travaux au début des années 1950 : l’injection à des animaux de laboratoire exerçait des effets marqués sur le taux de cholestérol hépatique et sur la cholestérolémie. Wattiaux montra que l’injection intra-péritonéale à des rats augmentait la proportion de phosphatase acide libre. En 1962, Wattiaux et Beaufay analysèrent par centrifugation en gradient de densité une fraction mitochondriale provenant de rats ayant, préalablement au sacrifice, reçu une injection de Triton WR1339. Ils eurent la surprise de constater que la densité d’équilibre des lysosomes était considérablement diminuée alors que celles des mitochondries et des organites à catalase n’étaient pas affectées. L’examen au microscope électronique de la fraction légère riche en phosphatase acide révélait la présence de lysosomes dilatés, au contenu clarifié, gorgés de Triton WR 1339.
Cette découverte permit la séparation partielle, par centrifugation en gradient de densité, des trois types d’organites de la fraction mitochondriale de rats injectés au Triton WR1339. La partie légère du gradient était enrichie en lysosomes modifiés (les « tritosomes ») ; les fractions lourdes contenait des organites entourés d’une membrane simple renfermant une structure cristalloïde ; ils étaient semblables aux « microbodies » de Rhodin. Une micrographie électronique, publiée en 1965 dans un article intitulé : « Combined Biochemical and Morphological Study of Particulate Fractions from Rat Liver », montre le nouvel organite ; la même année, il fut baptisé « peroxysome » par de Duve dans une communication au VIth Meeting of the American Society of Cell Biology. Sidney Goldfisher, un ancien collaborateur d’Alex Novikoff, mit au point une réaction cytochimique basée sur l’oxydation de diaminobenzidine par la catalase en présence de peroxyde d’hydrogène, pour révéler la présence de peroxysomes dans les cellules.
Une famille d’organites
Les peroxysomes sont présents dans toutes les cellules eucaryotes, des levures aux cellules humaines. Ils forment une famille d’organites comprenant les glyoxysomes. Leur équipement enzymatique varie selon le type de cellule et compte plus de 60 enzymes, parmi lesquels des oxydases catalysant l’oxydation (l’enlèvement d’hydrogène) de divers substrats, et transférant les électrons sur l’oxygène moléculaire :
![]()
Le peroxyde d’hydrogène produit est décomposé par la catalase peroxysomiale :
![]()
Les premiers substrats physiologiques caractérisés furent des acides aminés (de la série D), des α-hydroxy-acides et l’acide urique. Les peroxysomes sont des structures dynamiques à un double point de vue : ils sont en constant renouvellement, leur durée de vie n’excédant pas cinq jours et ils sont capables de déplacement le long du réseau de microtubules. Ils seraient connectés entre eux par de fins filaments.
Les mitochondries et les peroxysomes consomment 20% de l’oxygène utilisé par les hépatocytes. Dans ces deux organites, le principal accepteur d’électrons est l’oxygène moléculaire. Il est réduit en eau dans les mitochondries et en peroxyde d’hydrogène dans les peroxysomes. Ce produit est fortement toxique pour les cellules ; il donne naissance aux radicaux libres O2- (ion superoxyde) et OH– (radical hydroxyle), dont les électrons non appariés sont extrêmement réactifs et peuvent altérer irréversiblement les acides nucléiques, les protéines et les lipides cellulaires. Seamus V. Lennon (Department of Biology, Saint-Patrick’s College, Maynooth, Irlande) et Sten Orrenius (Karolinka Intitutet, Stockholm) ont, indépendamment, attiré l’attention sur le rôle du peroxyde d’hydrogène dans la mort cellulaire par apoptose (à faible concentration) et par nécrose (à forte concentration). C’est dire que la destruction de ce produit est cruciale pour la survie de la cellule. C’est pourquoi, malgré la diversité de leur équipement enzymatique selon les tissus, les peroxysomes renferment tous de la catalase. Cet enzyme ubiquitaire fut découvert en 1900 par le chimiste Oscar Loew (United States Department of Agriculture, Washington), un ancien élève de Justus von Liebig à l’université de Munich. La catalase consomme H2O2 au fur et à mesure de sa formation grâce à une capacité catalytique élevée (200.000 réactions catalysées par seconde à vitesse maximale), seulement limitée par la vitesse de diffusion du substrat. En 1936, David Keilin et E.F. Hartree (Molteno Institute, University of Cambridge) montrèrent que la catalase est une hémoprotéine pouvant agir comme une peroxydase.
Lorsque la concentration locale en peroxyde d’hydrogène est élevée, la catalase provoque la dismutation du peroxyde d’hydrogène en oxygène moléculaire et en eau :
![]()
Lorsque la concentration locale en peroxyde d’hydrogène est faible, la catalase oxyde des substrats donneurs d’électrons en utilisant le peroxyde d’hydrogène produit par les autres enzymes :
![]()
Cette réaction de peroxydation conférait aux peroxysomes un rôle dans la détoxification d’une série de substances introduites dans l’organisme par voies orale ou respiratoire dans l’organisme : phénols, alcool, méthanol, acide formique, formaldéhyde… Les réactions de peroxydation de la catalase sont surtout actives dans le foie et le rein des mammifères.
Fonction des peroxysomes
La β-oxydation est une voie catabolique qui permet aux cellules eucaryotes de récupérer l’énergie stockée dans les chaînes hydrocarbonées des acides gras ; elle constitue une source majeure d’énergie pour les êtres vivants. Elle fut, pour l’essentiel, découverte par Feodor F.K. Lynen (prix Nobel de physiologie ou médecine en 1964). Élève du chimiste Heinrich O. Wieland (prix Nobel de chimie en 1927), Lynen était à la tête du Max-Planck-Institut für Zellchemie, une structure spécialement créée pour lui à l’initiative d’Otto Warburg. Pour reconnaître sa contribution, la β-oxydation porte aussi l’appellation « hélice de Lynen », le terme « hélice » faisant référence au fait qu’à la suite des quatre réactions constituant ce processus on ne revient pas à la molécule initiale, comme dans le cycle des acides tricarboxyliques (molécule initiale : oxalo-acétate) ou dans le cycle de l’urée (ornithine), mais à un acide gras raccourci de deux unités carbonées. Les première et troisième réactions de l’hélice de Lynen sont des oxydations catalysées par deux déshydrogénases. La première déshydrogénation aboutit à la création d’une double liaison entre le carbone β et le carbone suivant, suivie de l’addition d’une molécule d’eau par une hydratase à la double liaison ainsi créée. Le β-hydroxyacyl est oxydé par une seconde déshydrogénase et une coupure par une thiolase libère un fragment en C2. Cette séquence de réactions se répète autant de fois qu’il est nécessaire pour dégrader complètement la molécule d’acyl-CoA en molécules d’acétyl-CoA (C2). Le catabolisme des acides gras se déroule en grande partie dans les mitochondries (voir chapitre « Mitochondries »).
Terrance G. Cooper et Harry Beevers (Department of Biological Sciences, Purdue University) montrèrent que dans l’endosperme des graines de Ricinus communis (castor bean) les enzymes de la β-oxydation sont localisés dans des granules : les glyoxysomes (voir plus loin). Le clofibrate est un agent thérapeutique qui modifie le métabolisme lipidique. L’examen au microscope électronique d’hépatocytes de rats auxquels cette substance a été administrée montre une prolifération des peroxysomes. Parallèlement, l’oxydation des acides gras est significativement augmentée. A la suite de ces observations, et considérant les similitudes biochimiques entre glyoxysomes et peroxysomes, Christian de Duve et Paul Lazarow (The Rockefeller University) établirent que l’oxydation du palmitoyl-CoA se déroule dans les peroxysomes d’hépatocytes de rats ayant reçu du clofibrate. En 1978, Lazarow montra qu’en plus de l’acyl CoA oxydase, ces organites contiennent les autres enzymes intervenant dans l’oxydation des acides gras en fonction de la longueur de leur chaîne hydrocarbonée : nulle avec l’acide butyrique (C4) comme substrat, la vitesse d’oxydation croissait avec l’acide octanoïque (C8), l’acide laurique (C12), l’acide palmitique (C16). Il en conclut que les peroxysomes oxydent préférentiellement les acides gras à longues chaînes.
Références: Cooper, TG, Beevers, H, β-Oxidation in glyoxysomes from castor bean endosperm (1969)
Lazarow PB, De Duve C, A fatty acyl-CoA oxidizing system in rat liver peroxisomes: enhancement by clofibrate, a hypolipidemic drug (1976)
Lazarow, PB, Rat liver peroxisomes catalyze the beta-oxidation of fatty acids (1978)
La β-oxydation dans les mitochondries présente des différences avec celle qui se déroule dans les peroxysomes : (i) dans les mitochondries, l’enzyme qui catalyse la première réaction du processus est une déshydrogénase qui transfère les électrons du FADH2 à la chaîne respiratoire ; dans les peroxysomes, c’est une oxydase qui transfère les électrons à l’oxygène avec production de peroxyde d’hydrogène (H2O2) ; hautement toxique pour la cellule, il est immédiatement détruit par la catalase sur son lieu de production. (ii) Dans les peroxysomes hépatiques la dégradation des acides gars s’arrête au stade C4, ou C3 si l’acide gras comprend un nombre impair de carbones (propionyl-CoA). (iii) L’acyl CoA-synthétase catalyse l’association au coenzyme A des acides gras présents dans le cytosol :
![]()
Les acides gras activés franchissent la membrane mitochondriale externe en s’associant à une petite molécule en C4 – la carnitine – portant un groupe carboxyl en C1 et une fonction ammonium quaternaire en C4. Pour passer de l’espace intermembranaire dans la matrice mitochondriale, où se déroule la β-oxydation, l’acyl-carnitine traverse la membrane mitochondriale interne en utilisant un transporteur spécifique, la carnitine/acylcarnitine tranlocase. Dans les peroxysomes, les acides gras franchissent la membrane en utilisant un transporteur spécifique sans association préalable à une molécule de carnitine. (iv) Les mitochondries oxydent les acides gras dont la chaîne n’excède pas 20 C. Les peroxysomes oxydent les acides gras à longues ou très longues chaînes (plus de 20 C), les acides gras à chaînes ramifiées et les acides gras dicarboxyliques à longue chaîne. Il en est de même pour la chaîne latérale des prostaglandines, thromboxanes et leucotriènes. Ces composés, regroupés sous le nom d’éicosanoïdes, sont des produits d’oxydation d’acides gras poly-insaturés en C18 (acide γ-linolénique) ou en C20 (acide arachidonique, acide eicosapentaénoïque). Prostaglandines, thromboxanes, et prostacyclines forment un sous-groupe au sein de cette famille de molécules complexes. Les leucotriènes sont les produits d’oxydation d’acide gras en C20 (acide arachidonique, acide dihomo-γ-linolénique, acide eicosapentaénoïque) par la 5-lipoxygénase. Ils appartiennent aussi à la famille des éicosanoïdes et se caractérisent par la présence dans leur formule chimique de trois doubles liaisons conjuguées :
![]()
Ces molécules sont des médiateurs d’une grande importance physiologique : les prostaglandines jouent un rôle essentiel dans les réactions inflammatoires, les thromboxanes, dans la coagulation sanguine, les leucotriènes, dans la contraction musculaire.
La β-oxydation dans les cellules végétales et chez les levures se déroule exclusivement dans les peroxysomes, à la différence de ce qui se passe dans les cellules animales. Elle prend toute son importance au moment de la germination des graines oléagineuses, en fournissant de l’acétyl CoA au cycle du glyoxylate au cours duquel sont synthétisés les glucides nécessaires à la croissance des plantes (voir paragraphe suivant). Les acides gras dont le Cβ porte un groupe méthyle ne sont pas dégradés par β oxydation. Ils empruntent une voie catabolique mineure : l’α-oxydation qui comporte trois étapes : (i) l’acyl-CoA est oxydé par une hydroxylase (dioxygénase) ; (ii) l’acide gras oxydé est clivé par une lyase en aldéhyde et formyl-CoA (en C1) ; (iii) une oxydase transforme l’aldéhyde en acide gras raccourci d’un carbone. Le groupe méthyle étant maintenant en position α, le catabolisme de l’acide gras peut se poursuivre par β oxydation. Le phytol, un alcool diterpénique présent chez les plantes et précurseur des vitamines D et E, est catabolisé par α-oxydation. Le déficit en hydroxylase catalysant la première étape de l’α-oxydation est la cause de la maladie de Refsum. Cette pathologie du groupe des leucodystrophies est caractérisée par une accumulation d’acide phytanique dans le sang et les tissus entraînant un déficit de l’audition, de la vision et des facultés intellectuelles.
Les plasmalogènes sont des alkényl-acyl-glycérophospholipides ou glycérophospholipides vinyl-éther. Je rappelle la formule de la liason vinyl-éther :
![]()
Le glycérol est substitué sur le carbone C1 (numérotation stéréospécifique sn-1) par un alcool gras (liaison vinyl-éther) ; sur C2 (sn-2), par un radical acyle (liaison ester) qui est souvent l’acide docosahexaénoïque (en C22) ou l’acide arachidonique (en C20). Le C3 (sn-3) du glycérol porte une phospho-choline (un alcool aminé abondant dans le muscle cardiaque), ou une phospho-éthanolamine (dans le cerveau), plus rarement un phospho-insositol ou une phospho-sérine (un acide aminé portant un groupe hydroxyle sur le Cβ). Comme le soulignent Narasimhan Nagan et Raphael A. Zoeller (Boston University School of Medicine), la présence de la liaison vinyl-éther confère aux plasmalogènes des propriétés anti-oxydantes vis-à-vis des espèces réactives de l’oxygène, renforcées par la présence sur le C2 du glycérol d’acide gras polyinsaturés oméga-3 et oméga-6.
Référence : N. Nagan, R. A. Zoeller, Plasmalogens: Biosynthesis and functions (2001).
Les plasmalogènes (1-alkyl-1-ényl-2-acyl glycérophosphoéthanolamines) sont très abondants dans le cerveau ; ils sont des constituants essentiels (près de 70%) des gaines de myéline entourant et protégeant les axones des cellules nerveuses. Dans le myocarde, ils représentent près de la moitié des phospholipides. Ils sont aussi présents dans certains cellules sanguines (polymorphonucléaires neutrophiles, macrophages, facteur d’agrégation des plaquettes sanguines) et dans la glande thyroïde. Ils sont peu abondants dans le foie et le rein. La biosynthèse des plasmalogènes est particulièrement active dans les cellules du système nerveux central. Au début des années 1980, il fut établi que la molécule de départ de la synthèse des glycérolipides, y compris ceux possédant une fonction vinyle-éther, est le dihydroxy acétone phosphate (DHAP), formé selon la réaction :
![]()
En 1982, Amiya K. Hajra et James E. Bishop (Department of biological chemistry, University of Michigan, Ann Arbor) montrèrent dans un article publié dans les Annals of the New York Academy of Sciences : « Glycerolipd biosynthesis in peroxysomes via the acyl dihydroxyacetone pathway », que les enzymes catalysant les deux premières réactions de la synthèse des plasmalogènes sont localisés dans les peroxysomes. Le DHAP, synthétisé dans le cytosol, franchit la membrane peroxysomiale et pénètre dans la matrice où il est acylé (liaison ester) par la DHAP acyl transférase, un enzyme caractérisé en 1981 par Lawrence M. Ballas et Robert M. Bell (Department of biochemistry, North Carolina State University) :
![]()
Au cours de l’étape suivante, la 1-alkyl-DHAP synthase catalyse la substitution de l’acide gras lié au DHAP par un alcool gras aliphatique à longue chaîne (liaison éther), synthétisé dans le cytosol et qui a franchi la membrane peroxysomiale :
![]()
L’exportation du 1-alkyl-DHAP vers le reticulum endoplasmique, où se déroulent les étapes suivantes de la synthèse des plasmalogènes, est catalysée par l’enzyme membranaire 1-alkyl-DHAP réductase :
![]()
Masanori Honsho, Shunsuke Asaoku et Yukio Fujiki (University of Hyogo, Kobe) ont montré que la production de plasmalogènes est régulée (down-regulation) par l’activité de l’acyl-CoA réductase 1 (1-alkyl-DHAP réductase). Ce complexe enzymatique membranaire est sensible à la teneur en plasmalogènes; en cas de déficience, son activité augmente; lorsque le taux de plasmalogènes revient à la normale, un mécanisme de rétroaction négative (negative feedback mechanism) se met en place et la vitesse de dégradation de l’enzyme s’accélère.
Référence: Masanori Honsho, Shunsuke Asaoku et Yukio Fujiki, Posttranslational regulation of fatty acyl-CoA reductase 1, Far1, controls ether glycerophospholipid synthesis
Le cholestérol est un précurseur des hormones stéroïdes et des acides biliaires, et un constituant des membranes, notamment de la membrane plasmique. La synthèse du cholestérol commence avec la formation d’hydroxy-méthyl-glutaryl-CoA dans le cytosol des hépatocytes et se poursuit dans le reticulum endoplasmique des cellules hépatiques et intestinales (pour 80%) et dans la matrice des peroxysomes (pour 20%) ; ce processus met en œuvre vingt enzymes différents, 18 molécules d’acétyl-CoA et 18 molécules d’ATP. Les premières étapes portent le nom de « voie du mévalonate » : l’acétoacétyl CoA thiolase (thiolase II) catalyse la condensation de deux molécules d’acétyl-CoA (C2) en acéto-acétyl-CoA (C4), qui est converti successivement en 3-hydroxy-3-méthylglutaryl-CoA par la 3-hydroxy-3-methylglutaryl CoA réductase, puis en mévalonate (C6) par la HMG-CoA réductase. La mévalonate (3 ou 5) kinase phosphoryle l’acide mévalonique en consommant une molécule d’ATP. Dans la suite des réactions, se forme un dérivé isoprénoïde en C5, l’isopentényl pyrophosphate ; c’est l’aboutissement de la voie du mévalonate ; la biosynthèse se poursuit par la condensation de six molécules d’isopentényl pyrophosphate en squalène (C30).
La bile est un liquide contenant – outre des électrolytes, des pigments (bilirubine, biliverdine), du cholestérol et des phospholipides – des sels biliaires formés par conjugaison d’acides biliaires avec les acides aminés glycine ou taurine. La majorité des acides biliaires sont des dérivés de l’acide cholique : acides taurocholique, glycocholique, chénodésoxycholique ; chez l’homme, les deux premiers représentent environ 80 % des acides biliaires. Ces stéroïdes en C24 sont des produits d’oxydation du cholestérol. Ils sont synthétisés par la voie dite classique, dont la première réaction est catalysée par la cholestérol 7α-hydoxylase :
![]()
et dont le produit final est le cholate, ou par la voie dite alternative, dont la première réaction est catalysée la stérol-27-hydroxylase :
![]()
et dont le produit final est le chénodésoxycholate. Ces deux hydroxylases sont des mono-oxygénases de la famille des cytochromes P450 localisées dans le reticulum endoplasmique. La conversion du cholestérol en acides biliaires met en jeu une cascade de vingt enzymes localisés principalement dans les mitochondries et les peroxysomes des cellules hépatiques ; chez l’homme, cette conversion consomme environ la moitié de la production journalière de cholestérol ; le reste est destiné à la membrane plasmique des cellules, à la synthèse des hormones stéroïdiennes et de la vitamine D. En 1991, Harald Osmundsen, Jon Bremer et Jan I. Pedersen (Department of Physiology and Biochemistn, UniversitY of Oslo) décrivirent l’oxydation de la chaîne latérale des précurseurs des acides biliaires, les acides dihydroxy- et trihydroxy-cholestanoïques, et leur conversion en cholyl-CoA. En 1980, Jan I. Pedersen et Johan Gustafsson (Chalmers University of Technology, Gothenburg) montrèrent que la synthèse de l’acide cholique se déroule dans les peroxysomes. Les acides biliaires, sous forme de sels, conjugués à la glycine (cholate) ou à la taurine (chénodésoxycholate) et fixation d’un ion Na+, jouent un rôle important dans la résorption du cholestérol et des graisses au niveau du tube digestif.
Références. Osmundsen H, Bremer J, Pedersen JI Metabolic aspects of peroxisomal β-oxydation (1991).
Pedersen JI, Gustafsson J, Conversion of 3 alpha, 7 alpha, 12 alpha-trihydroxy-beta-cholestanoic acid into cholic acid by rat liver peroxisomes (1980).
Enfin, les peroxysomes participent à la photorespiration au cours de laquelle les plantes consomment de l’oxygène et produisent du gaz carbonique ; ils renferment un enzyme impliqué dans ce processus, la glycolate oxydase qui catalyse la réaction :
![]()
(voir le paragraphe consacré au cycle oxydatif photosynthétique du carbone dans le chapitre « Chloroplastes »)
Glyoxysomes
Il existe un autre mécanisme, différent de la β-oxydation, qui permet aux cellules végétales et aux graines (riches en lipides) en cours de germination d’utiliser l’énergie des acides gras. Chez les bactéries, les champignons, les protistes et les plantes (mais pas chez les mammifères), le cycle du glyoxylate est une variante anabolique du cycle des acides tricarboxyliques. Il fut découvert par Hans Kornberg dans le laboratoire de Hans Krebs. Il porte le nom de l’un des intermédiaires du cycle, l’anion glyoxylate, un acide carboxylique en C2. La conversion des acides gras en sucres se fait par une suite de réactions au cours desquelles les groupes acétyl fournis par l’acétyl CoA sont condensés en succinate. Harry Beevers (University of California, Santa Cruz) démontra la présence dans les cellules d’endosperme de deux enzymes du cycle, la malate synthase et l’isocitrate lyase. Son élève, Bill Breidenbach, établit par centrifugation en gradient de densité la localisation de ces deux enzymes dans des organites plus denses que les mitochondries ; Beevers les nomma « glyoxysomes ». Ces organites sont présents dans les plantules, où la photosynthèse n’est pas encore fonctionnelle. Ils sont associés aux corpuscules lipidiques des graines dans lesquels ils puisent leurs substrats. La mobilisation rapide des graisses par les enzymes du cycle du glyoxylate fournit les glucides nécessaires à la croissance. James Hogg et Miklos Müller (The Rockefeller University) ont montré que les peroxysomes d’un protiste, Tetrahymena pyriformis, possèdent un cycle du glyoxylate incomplet ; l’absence de certains enzymes est compensée par une coopération métabolique entre peroxysomes et mitochondries.
Frederik R. Opperdoes et Piet Borst (Section for Medical Enzymology and Molecular Biology, Laboratory of Biochemistry, University of Amsterdam) identifièrent un organite de type peroxysomes chez un protozoaire parasite ; les « glycosomes » sont présents chez les protistes de l’ordre des Kinétoplastidés et chez les Diplonémidae (plancton des milieux aquatiques). Ils sont spécialisés dans la glycolyse, la biosynthèse des sucres nucléotidiques et, peut-être, d’autres voies métaboliques.
Références : Kornberg HL, Krebs HA. Synthesis of Cell Constituents from C2-Units by a Modified Tricarboxylic Acid Cycle (1957)
Opperdoes FR, et Borst P Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: the glycosome (1977)
Hydrogénosomes
Les hydrogénosomes ont été découverts dans les années 1970 par Donald G. Lindmark et Miklos Müller (The Rockefeller University) chez Trichomonas fœtus, un parasite du tube digestif et des organes génitaux. Ces « microcorpuscules » sont présents chez des parasites anaérobies comme les trichomonades, chez des ciliés et certains champignons. Leur taille est d’environ 1 micromètre et ils sont entourés d’une double membrane. Les hydrogénosomes s’apparentent aux mitochondries ; les cellules qui en possèdent sont dépourvues de mitochondries. Comme dans les mitochondries, leur membrane interne projette des crêtes dans la matrice ; à la différence des mitochondries, ils sont dépourvus de génome (sauf peut être chez le cilié Nyctotherus ovalis et le straménopile Blastocystis) et ne possèdent ni cytochromes ni oxydations phosphorylantes. Placés en conditions anaérobies, les hydrogénosomes se comportent comme des organites respiratoires produisant de l’hydrogène (H2) qu’ils oxydent en présence d’oxygène. Ils génèrent de l’ATP en oxydant du pyruvate ; à la différence des mitochondries, l’accepteur d’électrons n’est pas l’oxygène moléculaire mais le proton (H+) :
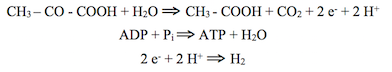
Les protons sont des accepteurs d’électrons seulement utilisés par un nombre restreint d’êtres vivants (anaérobies) probablement parce qu’ils sont peu efficaces ; la formation d’hydrogène requiert en effet un niveau de potentiel beaucoup plus élevé (50 kilocalories) que celui du couple eau/oxygène.
![]()
La production d’ATP dans les hydrogénosomes met en jeu moins d’enzymes que dans les mitochondries : une oxydoréductase (la pyruvate : ferrédoxine oxydoréductase), une hydrogénase, une transférase (acétate : succinate CoA transférase) et une kinase (succinate thiokinase). Les hydrogénosomes contiennent un enzyme essentiel pour la défense des cellules contre les effets dévastateurs des radicaux libres : la superoxyde dismutase, une métalloprotéine (à manganèse) qui catalyse la dismutation de l’ion superoxyde O2–. (cette écriture fait apparaître son caractère radicalaire) :
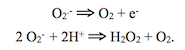
« Peroxisome proliferators »
En 1962 apparut dans l’arsenal thérapeutique une classe de substances dérivant de l’acide clofibrique. L’éthyl-p-chloro-phénoxy-isobutyrate (clofibrate) fut commercialisé en 1965 pour abaisser le taux de lipides et de cholestérol sanguins chez l’homme ; elle provoque cependant des effets secondaires indésirables parmi lesquels une hépatomégalie. En examinant au microscope électronique des coupes de foie de rat et de souris traités par le clofibrate, Donald J. Svoboda et Daniel L. Azarnoff (University of Kansas Medical Center) montrèrent que l’hépatomégalie s’accompagne d’une prolifération des microbodies dans les hépatocytes. L’augmentation du nombre de peroxysomes fut aussi observées par David E. Moody et Janardan K. Reddy (Northwestern University) dans les hépatocytes de rats traités par une variétés de dérivés d’acides dicarboxyliques : acides phtalique, adipique, sébacique… et chez la levure après administration de méthanol ou d’acide oléique. On observe parallèlement une augmentation du nombre de récepteurs nucléaires appelés Peroxisome Proliferator Activated Receptors (PPAR). Ces facteurs de transcription sont présents dans le foie (PPARa, d et g), les muscles (PPARd) et le tissu adipeux (PPARg) ; ils sont activés par l’augmentation du taux de lipides dans le sang ; leurs ligands sont des acides gras ou des dérivés d’acides gras. Les récepteurs PPAR activés s’associent aux récepteurs cytosoliques de l’acide rétinoïque en formant des hétérodimères qui franchissent l’enveloppe nucléaire et se fixent sur les régions régulatrices des gènes des enzymes du métabolisme des lipides et des hydrates de carbone. Ils jouent ainsi un rôle central dans l’homéostasie des lipides provenant de la digestion en adaptant leur catabolisme ou leur stockage en fonction des signaux fournis par les teneurs en acides gras ou en intermédiaires métaboliques.
Références : Svoboda D, Azarnoff DL. Response of hepatic microbodies to a hypolipidemic agent, ethyl chlorophenoxyisobutyrate (1966)
Moody, DE, Reddy, JK. Hepatic peroxisome (microbody) proliferation in rats fed plasticizers and related compounds (1978)
Maladies peroxysomiales : « Peroxisomes biogenesis disorders »
Les pathologies affectant les peroxysomes sont classées en deux types : celles découlant de la déficience d’un enzyme, et qui affectent une voie métabolique ; celles découlant d’un défaut de biogenèse, et qui affectent l’ensemble des voies métaboliques peroxysomiales. La gravité de ces affections donne la mesure de l’importance des fonctions cataboliques et biosynthétiques des peroxysomes. Les maladies génétiques par absence ou déficience ont été décrites dans les années 1960 ; elles entraînent le décès du patient. Le tissu nerveux est profondément altéré, notamment la substance blanche du cerveau. L’organisme ayant perdu la capacité de cataboliser un certain nombre de substances, qui s’accumulent dans le sang et les tissus. Le paradigme de ces maladies génétiques fut décrit en 1964 par le médecin H. Zellweger : le syndrome de Zellweger appartient au groupe de leucodystrophies que les cliniciens anglo-saxons appellent « Peroxisome Biogenesis Disorders ». Les nouveau-nés atteints présentent une hypotonie, des malformations osseuses au niveau du crane, des mains et des pieds, un retard de développement neurologique avec surdité et atteinte de la rétine, une hépatomégalie et des kystes rénaux ; ils décèdent dans les six premiers mois de leur existence. En 1973, en observant des cellules de patients présentant un syndrome de Zellweger, Sydney L. Goldfisher (Albert Einstein College of Medicine) découvrit qu’elles ne renfermaient pas de peroxysomes reconnaissables.
Dans l’adréno-leucodystrophie néonatale liée au chromosome X, le défaut génétique porte sur le transporteur des acides gras à très longue chaîne de la membrane des peroxysomes. Le défaut fut identifié en 1993 par Patrick Aubourg (Unité de neurologie pédiatrique, Institut National de la Santé et de la Recherche Médicale, Hôpital Saint-Vincent-de-Paul, Paris) et Jean-Louis Mandel (Laboratoire de génétique humaine, Faculté de médecine, Université Louis-Pasteur, Strasbourg). Les acides gras à très longue chaîne n’étant plus dégradés, ils s’accumulent dans le sang et les tissus provoquant chez les jeunes patients des troubles neurologiques, avec diminution des fonctions cognitives et motrices, perte de la vision et de l’audition.
Une autre forme de pathologie, appelée par certains cliniciens « Syndrome des peroxysomes vides » résulte d’un défaut d’adressage des protéines peroxysomiales. Les enzymes de la matrice sont synthétisés par les polysomes cytosoliques ; ils portent une séquence d’adressage (Peroxisomal-targeting Sequence, PTS) qui fut découverte au cours d’expériences avec la luciférase des cellules d’insecte. Cette séquence est le plus souvent située à l’extrémité C-terminale du polypeptide (PTS1), plus rarement à son extrémité N-terminale (PTS2). Elle est reconnue par un transporteur cytosolique, la peroxine PEX5 (Peroxisome Biogenesis Factor). Le complexe polypeptide-PTS1 – PEX5 se fixe sur un récepteur de la membrane des peroxysomes formé par l’association de protéines membranaires PEX, notamment PEX14, avant d’être transféré dans la matrice par un translocon. Un transporteur non-opérationnel par déficience de protéines PEX se traduit par un déficit en enzymes pouvant entraîner l’absence totale de peroxysomes et de β-oxydation, qui affecte la dégradation des acides gras à très longues chaînes, des acides gras branchés et de l’acide pristanique (un acide gras terpénoïde provenant de l’alimentation ou du lait maternel) ; l’augmentation de la concentration de ces molécules dans le sang et les tissus provoque des effets cliniques dramatiques chez les patients.
Les peroxysomes synthétisent les précurseurs du plasmalogène (1-alcylglycérone 3-phosphate). Les plasmalogènes sont des glycérophospholipides dans lesquels l’une des deux chaînes d’acides gras est remplacée par un alcool à longue chaîne lié au C1 du glycérol par une liaison éther (1-alcényl 2-acyl glycéro-phospholipides). Ce sont des constituants essentiels des gaines de myéline entourant et protégeant les axones des cellules nerveuses ; ils sont aussi présents dans les muscles en particulier dans le muscle cardiaque, où ils représentent près de la moitié des phospholipides. Dans les cas de déficience en enzymes des peroxysomes, la synthèse des plasmalogènes est perturbée ou supprimée.
- Il s’agit d’une « fausse latence » en ce sens qu’elle n’est pas due à l’imperméabilité de la membrane au substrat, comme dans le cas des lysosomes, mais au pouvoir catalytique (kcat) très élevé de la catalase par rapport à la vitesse de diffusion de l’eau oxygénée.
- Henri Beaufay et al., Tissue fractionation studies. 18. Resolution of mitochondrial fractions from rat liver into three distinct populations of cytoplasmic particles by means of density equilibration in various gradients. Biochemical Journal, 1964, 92, 184.