Anthonie van Leeuwenhoek a-t-il découvert le noyau des cellules ? Dans une lettre envoyée en 1702 à la Royal Society de Londres, il mentionna la présence d’un point lumineux au centre des globules rouges de poissons : « …a clear sort of light in the middle: ». Vingt ans plus tôt, il avait déjà observé la présence de globules dans les cellules sanguines de morue et de saumon mais, comme le fait remarquer Henry Harris dans « The Birth of the Cell » : « Leeuwenhoek saw globules everywhere. » Quoiqu’il en soit, la découverte du noyau des cellules végétales est attribuée au botaniste Robert Brown qui, au retour d’une mission d’exploration des côtes d’Australie, fut nommé Curator (conservateur) des collections botaniques du British Museum à Londres. Il publia un travail monumental sur la flore australienne et poursuivit des recherches sur les végétaux. En 1831, utilisant un microscope fabriqué à Londres par Banks & Sons et Dollon, il découvrit un organite sphérique au centre des cellules végétales. Le noyau des cellules animales fut observé quelques années plus tard, en 1835, par Rudolf F.J.H. Wagner, professeur de zoologie et d’anatomie comparée à l’université d’Erlangen.
Le noyau est le plus volumineux des organites subcellulaires ; d’un diamètre de 5 micromètres en moyenne, il est souvent sphérique (neurones), ovoïde (fibroblastes), ou plurilobé (leucocytes polynucléaires neutrophiles). Il disparaît au début de la division cellulaire et se reforme à la fin. Il y a généralement un noyau par cellule, deux dans certains hépatocytes ; il y en a plusieurs (syncitium) dans les rhabdomyocytes, les cellules contractiles des fibres musculaires striées résultant de la fusion du cytoplasme de plusieurs cellules. Le contenu du noyau – le nucléoplasme – est un gel aqueux renfermant, outre le matériel génétique, des nucléotides triphosphates et une variété de protéines ; il est séparé du cytoplasme par une enveloppe formée d’une membrane externe et d’une membrane interne séparées l’une de l’autre par un espace périnucléaire de 20 à 40 nm, en continuité avec la lumière du réticulum endoplasmique ; cet espace renferme des ions Ca2+ libérables dans le noyau par des canaux calciques membranaires. Les membranes externe et interne sont formées d’une bicouche phospholipidique et d’une variété de protéines membranaires (environ 80 protéines pour la membrane interne). La membrane externe porte des ribosomes. Membranes externe et interne sont percées de pores nucléaires, qui sont des structures de transit régulé entre cytoplasme et nucléoplasme. La face de la membrane interne en contact avec le nucléoplasme est tapissée d’un réseau fibrillaire dense d’une épaisseur de 10 à 20 nm, formé de protéines filamentaires codées par les gènes LMNA (lamines A, C, Ad10 et C2), LMNB 1 (lamine B1) et LMNB 2 (lamines B2 et B3). La teneur en ces diverses isoformes de lamines varie en fonction des phases du cycle cellulaire. Les lamines sont des constituants des filaments intermédiaires, présentes dans tous les types cellulaires. Elles remplissent un triple rôle : (i) partie intégrante du squelette, elles sont responsables de la forme du noyau et interviennent dans la distribution des pores nucléaires et les mouvements du noyau (caryocinèse) ; (ii) le réseau des lamines nucléaires est connecté au cytosquelette par l’intermédiaire d’un composant du complexe LINC (LInker of Nucleoskeleton and Cytoskeleton), le produit du gène Sad1-UNC-84 domain containing 1 (SUN1) ; (ii) elles servent de sites d’ancrage à l’hétérochromatine et aux télomères, soit directement, soit indirectement par l’intermédiaires de protéines : émerine, BAF (Barrier to Autointegration Factor) ou LBR (Lamin B Receptor) ; (iii) elles jouent un rôle dans la régulation de la transcription.
L’observation au microscope photonique permit de faire un premier inventaire du noyau. Au XIXe siècle, apparurent les dérivés de synthèse de l’aniline. Ces composés basophiles se fixent sur les acides nucléiques et colorent fortement la chromatine ; ils furent utilisés pour mettre au point de nouvelles techniques de coloration. Les chromosomes forment l’essentiel de la masse du nucléoplasme mais ils ne sont pas visibles dans la cellule en interphase (entre deux divisions cellulaires). En 1928, Emil Heitz distingua au sein du nucléoplasme des zones peu colorées, finement réticulées, qu’il appela « euchromatine » (c’est la partie active du génome, celle dont les gènes sont transcrits en ARN) et des amas foncés aux contours irréguliers situés à la périphérie et qu’il nomma « hétérochromatine » (c’est la portion du génome au repos plus des segments spécialisés des chromosomes appelées télomères et centromères). L’emploi de techniques d’hybridation in situ couplées à la microscopie à fluorescence permit de montrer que le nucléoplasme est une structure dynamique et organisée dans laquelle les chromosomes individuels ne sont pas répartis de manière aléatoire mais occupent des territoires distincts, séparés par un réseau canaliculaire (domaine inter-chromosomique) dans lequel circulent les facteurs de régulation. Les chromosomes de petite taille ou riches en gènes sont préférentiellement localisés dans la partie centrale du nucléoplasme ; ceux de grande taille ou pauvres en gènes, à la périphérie, ancrés à la lamina par la protéine II (Lamin associated protein II) et le récepteur de la lamine B.
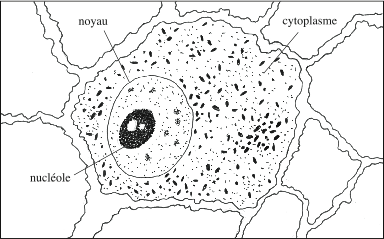 |
| Cellule de tumeur ascitique observée au microscope à contraste de phase. Le noyau est entouré par le cytoplasme, contenant diverses inclusions. La cellule est bordée par la membrane plasmique. La microscopie à contraste de phase permet l’examen de tissus non fixés et non colorés. |
Chez les mammifères et les oiseaux, le nucléoplasme renferme une variété de structures dynamiques appelés corps nucléaires, dont le nombre peut varier en fonction des conditions de stress auxquelles sont soumises les cellules. On en a identifié une dizaine. Dans ce chapitre, je ne décrirai que les mieux connues. Dépourvus de membrane, les corps nucléaires maintiennent leur intégrité en renouvelant leurs constituants par une biogénèse très active et des échanges de matériel avec le nucléoplasme environnant. La récupération de fluorescence après photo-blanchiment (Fluorescence Recovery after Photobleaching, qui mesure la vitesse de diffusion de molécules fluorescentes) a permis de mettre en évidence les processus dynamiques d’auto-association protéines-protéines et protéines-ARN au sein des corps nucléaires, par exemple en réponse à des stimulations externes (infection virale) ou internes à la cellule.
L’inclusion la plus volumineuse est le nucléole, une masse hétérogène aux contours irréguliers (voir plus loin). Les corps de Cajal (1 à 3 exemplaires par noyau) furent décrits en 1903 par l’histologiste Santiago Ramon y Cajal (prix Nobel de physiologie ou médecine en 1906). Ils prennent la forme de petites masses arrondies de 200 à 600 nanomètres de diamètre. Ils sont présents dans le noyau des neurones – où Cajal les a découverts ; ils le sont moins ou sont absents dans le noyau d’autres types cellulaires. Les corps de Cajal renferment des fils enroulés, visibles à l’examen au microscope électronique, d’où l’appellation de « coiled bodies ». Ils sont formés de protéines, dont la coïline-p80, et d’ARN et sont impliqués dans l’assemblage et la maturation de petites ribonucléoprotéines nucléaires (Small Nuclear Ribonucleoproteins) et nucléolaires (Small Nucleolar Ribonucleoproteins). Les Corps de Cajal jouent aussi un rôle dans l’assemblage de la télomérase et dans l’homéostasie de la longueur des télomères (voir plus loin).
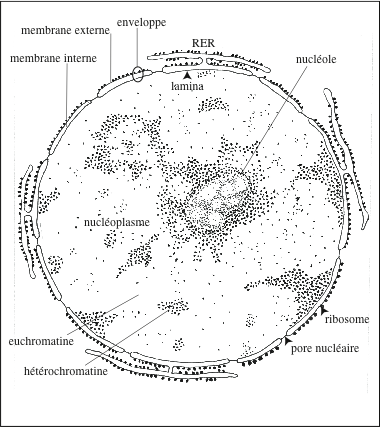 |
| Noyau d’une cellule en interphase, observé au microscope électronique. |
Les speckles (Nuclear speckles, 20 à 50 exemplaires par noyau) apparaissent au microscope à fluorescence sous forme de petites masses brillantes (speckles signifie « mouchetures ») de forme irrégulière. L’examen au microscope électronique révèle la présence de granules de 20 à 25 nm associés en grappes de 80 à 180 nm. Leur localisation dans le domaine inter-chromosomique du nucléoplasme leur a valu l’appellation d’Interchromatin granule cluster. Ce domaine, riche en fibrilles péri-chromatiniennes de 3 à 5 nm de diamètre, est une région de transcription active. Les Speckles sont des sites de stockage des facteurs d’épissage des pré-ARNm : SRSF1 (Serine/Arginine Rich Splicing Factor 1,) et ASF1/SF2. Les paraspeckles ont été découverts en 2002 par Archa H. Fox (Western Australia Institue for Medical Reaserch). Dans les cellules HeLa, l’examen au microscope électronique ou par fluorescence révèle la présence de 13 à 17 structures de forme irrégulière (0.5 à 1 mm de diamètre) par noyau. Les paraspeckles sont présents dans les cellules de mammifères et dans les lignées cellulaires transformées, principalement dans l’espace inter-chromatinien, à proximité des speckles. Ces complexes ARN/protéines se forment autour d’un ARN long non-codant (Nuclear Enriched Abundant Transcript 1, NEAT1) servant de centre de nucléation, et de protéines contenant des séquences de liaison à l’ARN, comme la Drosophila Behaviour/Human Splicing, DBHS. Au cours du cycle cellulaire, les paraspeckles sont visibles pendant l’interphase et la mitose, sauf pendant la télophase (l’ARN polymérase II est inactive pendant la télophase). La présence de paraspeckles est liée à l’activité de cet enzyme. Les paraspeckles sont des structures dynamiques contrôlant l’expression des gènes en séquestrant certains ARN dans l’espace inter-chromatinien. Le mécanisme de rétention d’ARN hyper-édités a été particulièrement étudié par le groupe de David L. Spector (Cold Spring Habour Laboratory). Pour rappel, après transcription les pré-ARN subissent une phase dite d’édition au cours de laquelle une adénosine est convertie en inosine par désamination hydrolytique. André P. Gerber et Walter Keller (Department of Cell Biology, Biozentrum, Universität Basel) furent des pionniers dans la découverte du processus d’édition en découvrant en 2001, l’adénosine désaminase spécifique de l’ARNt. Les paraspeckles séquestrent les molécules d’ARN qui ne sont pas immédiatement nécessaires dans le cytoplasme pour la synthèse protéique mais qui doivent être rapidement mobilisables en cas de stress.
Les corps nucléaires PML (10 à 30 exemplaires par noyau) tirent leur nom de la protéine suppresseur de tumeur ProMyelocytic Leukemia protein, identifiée en 1996 par Fabrizzio Grignani et ses associés (Università degli Studi di Milano) chez des patients atteints de leucémie promyélocytaire aigüe. La protéine PML joue le rôle de facteur de nucléation sur lequel s’agrègent d’autres protéines pour former les structures sphériques des corps nucléaires PML. Ceux-ci jouerait un rôle dans le contrôle de la transcription et de l’homéostasie cellulaire. Le nombre de corps nucléaires PML augmente chez les cellules soumises à un stress oxydatif.
Nucléole, une usine à ribonucléoprotéines
En 1781, le naturaliste Felice G.F. Fontana (université de Pise) rapporta la présence d’une structure arrondie à l’intérieur du noyau de cellules de mucus de peau d’anguille. Utilisant de nouvelles techniques de coloration, Rudolph F.J.H. Wagner (Friedrich-Alexander Universität, Erlangen) confirma en 1835 la présence de cet organite, qui fut nommé « nucléole » en 1836, par Gabriel G. Valentin (Université de Berne) puis, en 1840, par William Bowman, l’histologiste qui découvrit dans le rein les capsules qui portent son nom. Le réactif de Feulgen (à base de fuschine), qui colore la chromatine nucléaire, ne colore pas le nucléole. Cet organite, aussi appelé « tache germinative », est visible au microscope à contraste de phase sur des cellules en culture non fixées, ce qui a permis à Gérard-Edouard Balbiani, professeur d’embryologie au Collège de France, d’observer son caractère dynamique et les mouvements qui se déroulent en son sein. A la mitose, le nucléole disparaît en même temps que le noyau.
Référence : Balbiani EG Sur l’origine des cellules du follicule et du noyau vitellin de l’œuf chez les Géophiles (1864)
Il n’y a pas de nucléole ou de structure équivalente chez les procaryotes. Le matériel de choix pour l’étude de la structure et de la physiologie de cet organite est l’ovocyte, une cellule de l’ovaire qui se différencie en ovule. Les ovocytes d’araignées, utilisés par Balbiani comme matériel expérimental, ont des nucléoles bien développés. L’emploi de techniques histochimiques fournit peu de résultats utiles sur leur composition biochimique. En 1936, Torbjörn O. Caspersson (Kemiska Institutionen, Karolinska Institutet, Stockholm) effectua sa thèse de doctorat sur la répartition des acides nucléiques dans le noyau. Il nota deux faits : (i) l’examen par microspectrophotométrie dans l’ultraviolet détecte un pic d’absorption caractéristique des acides nucléiques dans le nucléole de noyaux isolés ; (ii) le nucléole ne réagissant que faiblement à la réaction de Feulgen, le pic d’absorption devait donc être celui d’ARN. Caspersson émit la supposition que le nucléole est le lieu de synthèse des ribonucléoprotéines (exact) et des protéines cytoplasmiques (faux). En 1937, Jack Schultz, un élève de Thomas Hunt Morgan, rejoignit le laboratoire de Caspersson. Ensemble, ils précisèrent la répartition des deux types d’acides nucléiques dans le noyau : l’ADN dans les chromosomes et l’ARN dans le nucléole. Ils confirmèrent que le cytoplasme renferme aussi de l’ARN et qu’il doit exister une relation métabolique entre ADN et ARN : la basophilie du cytoplasme augmente nettement dans les cellules qui se divisent activement.
Référence : Caspersson TO, Schultz J Ribonucleic acids in both nucleus and cytoplasm, and the function of the nucleolus (1940).
L’importance du nucléole a été mise en lumière au cours des années 1960. Donald D. Brown (Department of Embryology, Carnegie Institution of Washington, Baltimore) et John B. Gurdon (Department of Zoology, Oxford, prix Nobel de physiologie ou médecine en 2012) ont montré que les œufs d’un mutant de la grenouille sud-africaine Xenopus laevis, dépourvus de nucléole, ne sont pas viables ; la synthèse d’ARN ribosomal est abolie. Max L. Birnstiel et Hugh Wallace (Medical Research Council Epigenetics Research Group, Edinburgh), par une série de centrifugations isopycniques, parvinrent à isoler les gènes codant pour l’ARNr de Xenopus laevis. C’était la première fois qu’un gène était isolé ! La portion de la chromatine présente dans le nucléole (5% de la masse du nucléole) est localisée dans la zone fibrillaire, au centre de la structure. Utilisant la même technique de centrifugation, Oscar L. Miller, « the magician of molecular biology », et sa technicienne, Barbara R. Beaty (Oak Ridge National Laboratory) isolèrent des nucléoles d’oocytes de Xenopus laevis ; ils leur appliquèrent une technique d’étalement de la chromatine qu’ils avaient mise au point. Le résultat de l’examen au microscope électronique en transmission était spectaculaire : on a pu « voir », pour la première fois, la transcription de la chromatine en action. Le matériel nucléolaire décompacté apparaît sous forme de longues fibres d’ADNr (sensibles à la DNase) – les « arbres de Noël » ; le tronc de l’arbre est constitué par les espaceurs non transcrits ; les branches qui s’y attachent sont des filaments d’ARNr de longueur croissante au fur et à mesure de leur transcription ; on voit également des« boules » de nature protéique ; les grains raccordant les branches au tronc sont les ARN polymérases I.
Références : Brown DD, Gurdon JB Absence of Ribosomal RNA Synthesis in the Anucleolate Mutant of Xenopus laevis (1964)
Birnstiel ML, Wallace H, Sirlin JL, Fischberg M Localization of the ribosomal DNA complements in the nucleolar organizer region of Xenopus laevis (1966)
Wallace H, Birnstiel ML Ribosomal cistrons and the nucleolar organizer (1966)
Miller O, Beatty B Visualization of nucleolar genes (1969)
Jean Brachet, un pionnier de l’ « embryologie chimique », et Walter Vincent (Laboratoire de morphologie animale, Université libre de Bruxelles) isolèrent le nucléole d’hépatocytes de rat et d’ovocytes d’étoile de mer. Alors que dans la plupart des autres cellules, on dénombre de un à quatre nucléoles, selon les phases du cycle cellulaire, les ovocytes d’Amphibiens renferment de nombreux nucléoles. En 1940, Brachet démontra que le nucléole contient de l’ARN : la basophilie de cette structure disparait après traitement par la ribonucléase. Le matériel nucléolaire n’est pas réparti de manière uniforme : la zone granulaire est sensible à la ribonucléase alors la pepsine digère préférentiellement les autres parties du nucléole (son contenu en protéines représente 85% de sa masse). La présence d’ARN dans le nucléole d’ovocytes d’étoile de mer fut confirmée par Vincent (Zoology Laboratory, University of Pennsylvania).
La structure fine du nucléole fut définitivement établie par l’examen de coupes de tissus au microscope électronique. et Wilhelm Bernhard, Françoise Haguenau et Charles Oberling (Institut de recherches sur le cancer, Villejuif) décrivirent la structure en éponge du nucléole, l’absence de membrane limitante et la présence de fibrilles et de granules réparties en trois régions : une région riche en fibrilles de 5 à 8 nm de diamètre, de densité variable, qui se différencie en « centres fibrillaires » et « composant fibrillaire dense » ; une région riche en granules de 15 à 20 nm de diamètre : le « composant granulaire ».
Références : Brachet J La localisation de I’acide thymonucléique pendant I’oogenèse et la maturation chez les Amphibiens (1940)
Brachet J La localisation des acides pentosenucléiques dans les tissus animaux et les oeufs d’Amphibiens en voie de développement (1941)
Brachet J Ribonucleic acids and the synthesis of cellular proteins (1960)
Bernhard W, Haguenau F, Oberling C L’ultrastructure du nucléole de quelques cellules animales revélée par le microscope électronique (1952)
Bernhard W, Haguenau F, Gautier A, Oberling C La structure submicroscopique des éléments basophiles cytoplasmiques dans le foie, le pancréas et les glandes salivaires (1952)
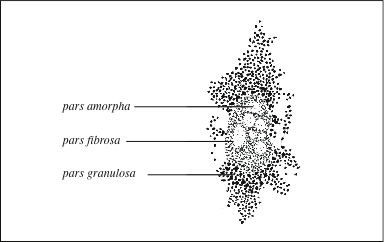 |
| Morphologie du nucléole chez les eucaryotes supérieurs. Chez les eucaryotes supérieurs, le nucléole est une structure dépourvue de membrane limitante ; ses trois composants sont : (i) en position centrale, le centre fibrillaire dense (anciennement pars fibrosa), avec les gènes transcrivant l’ARN ribosomal, l’ARN polymérase I et les facteurs de transcription ; (ii) la zone granulaire (pars granulosa) d’assemblage des sous-unités ribosomales 40S et 60S ; (iii) un centre fibrillaire regroupant cinq paires de chromosomes avec les régions NOR (Nucleolar Organizing Region) et les gènes répétés en tandem codant l’ARNr 45S. |
En traitant une préparation enrichie en nucléoles par un détergent (désoxycholate de sodium), Max L. Birnstiel et Margaret I.H. Chipchase (Division of Biology, California Institute of Technology) isolèrent des ribonucléoprotéines de coefficient de sédimentation 38S, 60S, et 81S et de tailles 20, 28, et 36,5 nm – soit la taille des ribosomes cytoplasmiques ou de leurs sous-unités – et des ARNr 18S et 28S. Les travaux Max Birnstiel et Hugh Wallace (Medical Research Council Epigenetics Research Group, Edinburgh) et de Donald D. Brown (Department of Embryology, Carnegie Institution of Washington, Baltimore) démontrèrent que les gènes codant pour les ARNr 18S et 28S sont organisés en tandems tête-bêche, répétés des centaines de fois dans le génome.
Référence : Birnstiel ML, Chipchase MIH The nucleolus, a source of ribosomes (1963)
En constatant la faible réactivité du nucléole à la coloration de Feulgen, les expérimentateurs avaient conclu qu’il ne contient pas ou peu d’ADN. En 1931, Emil Heitz (département de Botanique, Universität Hambourg) – l’inventeur des termes « euchromatine » et « hétérochromatine » – mit en évidence le contact étroit entre certains chromosomes et le nucléole de cellules végétales. En observant des chromosomes de plantes irradiées aux rayons X dans le laboratoire de Thomas Hunt Morgan (California Institute of Technology), la cytogénéticienne Barbara McClintock (Cold Spring Harbor Laboratory) découvrit qu’une région particulière du chromosome se positionne au contact du nucléole (1934) ; elle la baptisa « organisateur nucléolaire » (Nucleolar Organizing Body). Pour démontrer le rôle de cette région, aujourd’hui dénommée Nucleolus Organizer Region (NOR) , des transgènes codant pour des pré-ARNr furent insérés dans des chromosomes polythènes isolés (visibles au microscope optique) et incubés dans un milieu de transcription : des nucléoles se formèrent spontanément. Dans la cellule, les NOR sont concentrées dans les centres fibrillaires ; ces locis hébergent des centaines de gènes d’ADNr, codant pour les ARNr 5.8S, 18S et 28S (chez l’homme). Trois décades après leur publication (dans les décennies 1940-1950) la communauté scientifique prit conscience de l’importance des découvertes de Barbara McClintock. Ses travaux sur la cytogénétique du maïs et la découverte des transposons lui valurent l’attribution du prix Nobel de médecine ou physiologie 1983. Fait remarquable dans les annales du Comité Nobel, elle fut la seule lauréate ; elle avait 81 ans.
Pour assurer les besoins de la cellule en ribosomes fonctionnels, surtout en condition de stress, vitesse de synthèse et d’assemblage sont impératives ; les gènes d’ADNr sont localisés sur un petit nombre de chromosomes – cinq chez l’homme, pour 400 copies – et regroupés en opérons (clusters) ; la rapidité de transcription par l’ARN polymérase I est favorisée par la disposition des gènes d’ADNr en tandem. La transcription des gènes d’ADNr par l’ARN polymérase I se déroule à la limite entre centres fibrillaire et composant fibrillaire dense. Le long précurseur polycistronique (pré-ARNr 47S chez l’homme) subit plusieurs étapes de maturation dans le compartiment fibrillaire dense, où sont concentrées fibrillarine, petites ribonucléoprotéines nucléolaires (Small Nucleolar Ribonucleoproteins, snoARN) et une variété d’enzymes : méthyl transférases, isomérases, nucléases… Les ARNr subissent des modification consistant en méthylations et pseudo-uridylations. L’excision des exons des ARNr primaires – les séquences ETS (External Transcribed Sequence) et ITS (Internal Transcribed Sequence) – fait intervenir divers facteurs : snoARN, exonucléases, hélicases… et aboutit au découpage du pré-ARNr 47S en ARNr-5,8S, ARNr-18S et ARNr-28S. L’ARNr 5S, codé par des gènes répétés situés à proximité des centromères des chromosomes, sont transcrits à la périphérie du nucléole par l’ARN polymérase III. L’ARNr 5S se lie d’abord à deux protéines ribosomiales puis, dans la zone granulaire, aux autres protéines ribosomiales synthétisées dans le cytosol par l’ARN polymérase II, et importées via les pores nucléaires ; les sous-unités ribosomiales formées sont exportées dans le cytoplasme via les pores nucléaires. Les ribosomes fonctionnels se forment par association des grande et petite sous-unités ribosomiales entre elles et avec une molécule d’ARNm.
Le concept de nucléole multifonctionnel fut introduit en 1998 par Thoru Pederson (Department of Biochemistry and Molecular Biotechnology, University of Massachusetts). Si la fonction principale du nucléole est de fabriquer des ribosomes, il est aussi le site de biosynthèse d’autres ribonucléoprotéines.
Référence : Pederson T The plurifunctional nucleolus (1998)
La ribonucléase P est un ribozyme ; c’est même le premier ribozyme à avoir été identifié , ce qui valut à Sidney Altman (Medical Research Council Laboratory of Molecular Biology, Cambridge) de recevoir le prix Nobel de chimie 1989. L’ARNase P est une ribonucléoprotéine composée de l’ARN M1 (317 bases) et de la protéine C5 (119 acides aminés). Un traitement par une ribonucléase fait disparaître l’activité catalytique ce qui prouve que le site catalytique est associé à l’ARN M1. En 1983, Altman décrivit l’activité nucléolytique de l’ARNase P sur l’extrémité 5’ des pré-ARNt. En éliminant une séquence additionnelle située à l’extrémité 5′ du pré-ARNt, elle joue un rôle déterminant impliquée dans la maturation des ARN de transfert en ARNt fonctionnels.
Références : Stark BC, Kole R, Bowma EJ, Altman S Ribonuclease P: an enzyme with an essential RNA component (1978)
Kole R, Altman S Properties of the protein component of ribonuclease P from Escherichia coli (1980)
Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme (1983)
Biogenèse de la télomérase. Les télomères sont des séquences d’ADN répétitives, non-codantes, situées à l’extrémité des chromosomes. Leur importance fut mise en évidence dans les années 1980 par Jack W. Szostak (Department of Molecular Biology, Massachusetts General Hospital). Ces séquences particulières ne sont pas synthétisées par la polymérase responsable de la réplication de l’ADN chromosomal (ADN polymérase II) mais par la télomérase (TElomerase Reverse Transcriptase, TERT). Pour des raisons mécaniques, à chaque cycle de réplication, la longueur des chromosomes diminue à leurs extrémités. La fonction de la télomérase est de restaurer la partie manquante de façon à maintenir intacte la longueur des chromosomes en ajoutant une séquence répétée de désoxyribonucléotides (télomère) à l’extrémité des chromosomes linéaires. Elizabeth Blackburn (University of California, Berkeley) détecta la présence à l’extrémité des chromosomes de Tetrahymena thermophila de séquences d’ADN répétitives (- CCCCAA -) non-codantes. En 1980, Blackburn et Jack W. Szostak (Department of Molecular Biology, Massachusetts General Hospital) montrèrent que lorsque ces séquences sont greffées à l’extrémité de minichromosomes synthétiques et que ceux-ci sont injectés dans des cellules de Saccharomyces cerevisiae, ils ne subissent pas de dégradation, ce qui n’est pas le cas de minichromosomes dépouvus de séquences – CCCCAA – (Szostak était un pionnier de la synthèse de minichromosomes artificiels de levure). Szostak et Blackburn ont montré que l’ADN linéaire de plasmides de levure est protégé de la dégradation si on lui greffe des extrémités de chromosomes de Tetrahymena. Des minichromosomes artificiels de levure, introduits dans des cellules de levure, ne sont répliqués que si on leur ajoute des télomères aux deux extrémités. A la suite du séquençage de ces fragments d’ADN, l’enzyme responsable de leur addition aux extrémités terminales des chromosomes, la télomérase, fut découverte en 1984 dans des extraits de Tetrahymena thermophila par Carol Greider, l’assistante d’Elizabeth Blackburn. Cette ribonuléoprotéine est formée de l’association d’une transcriptase réverse, l’ADN polymérase TERT (synthétisée dans le cytosol), et d’un ARN de petite taille, le TElomerase Rna Component (TERC) . La présence de ces deux constituants dans les corps de Cajal de cellules cancéreuses humaines en culture, a conduit à attribuer à ces corps nucléaires un rôle dans l’assemblage et/ou la maturation de la télomérase et dans l’homéostasie de l’allongement des télomères. Au cours de la phase S du cycle cellulaire – pendant laquelle se produit l’élongation des télomères – les Corps de Cajal s’associent aux extrémités des chromosomes ; l’ARNTERC sert de matrice à l’ADN polymérase TERT en s’appariant aux séquences complémentaires des télomères ; l’enzyme synthétise et insère les séquences d’ADN répétitives perdues à chaque cycle de réplication. La télomérase est active dans les cellules germinales, embryonnaires ou fœtales et peu active dans les cellules somatiques. Elizabeth H. Blackburn, Carol W. Greider et Jack W. Szostak se partagèrent le prix Nobel de physiologie ou médecine 2009.
Références : Szostak JW, Blackburn EH Cloning yeast telomeres on linear plasmid vectors (1982)
Shampay J, Szostak JW, Blackburn EH DNA sequences of telomeres maintained in yeast (1984)
Greider CW, Blackburn EH Identification of a specific telomere terminal transferase activity in Tetrahymena extracts (1985)
La particule de reconnaissance du signal (Signal Recognition Particle, SRP) reconnait, comme son nom l’indique, le peptide signal, découvert en 1972 par César Milstein et ses associés (Department of Biochemistry, Cambridge University) à l’extrémité amino-terminale du précurseur de la chaîne légère d’immunoglobuline IgG. La particule fut isolée en 1980 dans le laboratoire de Günter Blobel (The Rockefeller University) par Peter Walter et Ibrahim M. Ibrahimi. Chez les eucaryotes, cette machine moléculaire est formée par l’association d’un ARN 7S (+/- 300 nucléotides) et de six polypeptides nommés d’après leur poids moléculaire en kDa : 72, 68, 54, 19, 14 et 9 kDa. Les cellules fabriquent en permanence des protéines dont certaines deviendront des résidentes (protéines domestiques) et d’autres seront exportées à l’extérieur de la cellule (protéines sécrétoires). Le choix d’entrer ou pas dans la voie sécrétoire est déterminé par la présence ou l’absence d’un peptide signal à l’extrémité N-terminale du polypeptide en cours de synthèse par les ribosomes cytosoliques. La particule de reconnaissance du signal, assemblée dans le nucléole et exportée dans le cytosol, joue un rôle déterminant dans l’adressage des protéines. L’hétérodimère SRP9/SRP 14 bloque l’élongation du polypeptide en cours de traduction ; SRP19 et la GTPase SRP54interagissent avec le peptide signal émergeant de la grosse sous-unité du ribosome ; SRP68 et SRP72 sont impliqués dans l’interaction de la particule de reconnaissance du signal avec son récepteur sur la face cytosolique de la membrane du reticulum endoplasmique. Cette double reconnaissance du peptide signal et du récepteur assure l’accostage et l’arrimage de l’ensemble ribosome – polypeptide naissant à la membrane du reticulum endoplasmique, qui est la porte d’entrée de la voie sécrétoire.
Références : Walter P, Blobel G Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum (1980)
Walter P, Ibrahimi I, Blobel G Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synyhesizing secretory protein (1981)
Walter P, Blobel G Translocation of proteins across the endoplasmic reticulum. II. Signal recognition protein (SRP) mediates the selective binding to microsomal membranes of in-vitro-assembled polysomes synthesizing secretory protein (1981)
Walter P, Blobel G Translocation of proteins across the endoplasmic reticulum. III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes (1981)
Walter P, Blobel G Signal recognition particle contains a 7S RNA essential for protein translocation across the endoplasmic reticulum (1982)
Petits ARN nucléolaires et maturation des ARNr. Deux types d’ARN coexistent dans les cellules : les ARN codants (ARNm) et les ARN non codants (ARNnc). Ces derniers sont impliqués dans des processus biologiques comme la régulation post-transcriptionnelle de l’expression du matériel génétique ou le développement de l’organisme. La taille des ARNnc est variable : de milliers de bases à une vingtaine (microARN de 21-23 bases). Les petits ARN nucléolaires (Small NucleOlar RNA, snoARN) sont présents dans les petites ribonucléoprotéines nucléolaires (Small NucleOlar RiboNucleoProteins, snoRNP). Les snoARN sont impliqués dans la modification des ARNr et d’autres petits ARN par voie chimique (méthylation, pseudouridylation) et dans le clivage des ARN ribosomiques (ARNr). En s’appariant avec les bases complémentaires de l’ARNr cible, les ARNsno à boîtes C et D (bien conservées) assurent la spécificité de la 2′-O-méthylation des riboses de l’ARNr par la fibrillarine. La zone non-appariée de la structure en double épingle à cheveux des ARNsno à boîte H/ACA se lie se lie aux bases complémentaires de l’ARNr cible assurent la spécificité de la conversion des uridines en pseudo uridines par la dyskérine.
Régulation de la biogenèse des ribosomes et de la progression du cycle cellulaire. Le nucléole est le site de réactions de sumoylation ; il s’agit d’une modification post-traductionnelle réversible où la petite protéine SUMO est liée de manière covalente à des protéines cibles. La découverte des petites protéines SUMO (Small Ubiquitin-related MOdifiers), dans les années 1996-1997, et de la voie de la SUMOylation, est attribuée à deux groupes : celui de Frauke Melchior, d’abord lorsqu’elle était étudiante postdoctorale dans le laboratoire de Larry Gerace (The Scripps Research Institute, San Diego), puis dans son laboratoire (Zentrum für Molekulare Biology, Universität Heidelberg), et celui de Michael J. Matunis (Johns Hopkins University) associé à Günter Blobel (Rockefeller University, Howard Hughes Medical Institute, New York). Dans les années 1990, au cours des travaux de son groupe sur le tri et l’adressage des protéines cellulaires, Blobel avait été amené à s’intéresser au rôle de protéines régulatrices des pores nucléaires comme RanGAP1 (Ran GTPase-Activating Protein 1).
Les pores nucléaires sont des complexes protéiques qui régulent le transport bidirectionnel de matériel (ARNm, protéines, polymérases…) entre le nucléoplasme et le cytoplasme, en franchissant l’enveloppe nucléaire. La petite protéine G régulatrice du transport nucléo-cytoplasmique GTPase Ran (Ras-like nuclear GTPase) existe sous deux formes : RAN-GTP et RAN-GDP. La conversion d’une forme à l’autre est catalysée par RanGAP1, une protéine qui existe aussi sous deux formes : la forme cytosolique et une forme plus lourde, liée aux fibrilles des pores nucléaires. Frauke Melchior a montré que ce changement de localisation et de poids moléculaire est dû au fait qu’une petite protéine SUMO (101 acides aminés) – apparentée à l’ubiquitine – est liée de manière covalente à RanGAP1. Cette réaction est catalysée par des ligases ; Melchior a identifié la première de ces ligases : laSUMO E3 ligase.
Références : Mahajan R, Delphin C, Guan T, Gerace L, Melchior F A small ubiquitin related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2 (1997)
Pichler A, Gast A, Seeler JS, Dejean A, Melchior F The nucleoporin RanBP2 is a SUMO1 E3 ligase (2002)
Barysch SV, Dittner C, Flotho A, Becker J, Melchior F Identification and analysis of endogenous SUMO1 and SUMO2/3 targets in mammalian cells and tissues using monoclonal antibodies (2014)
Michael J. Matunis, Elias Coutavas et Günter Blobel montrèrent que RanGAP1 est localisée dans le cytosol ; après avoir établi une liaison covalente avec SUMO, RanGAP1 change de poids moléculaire (90 kDa au lieu de 70) et de localisation en s’associant à la protéine du complexe du pore nucléaire RanBP2. Pour Matunis, SUMO peut être apparentée à une chaperone : elle empêche l’agrégation des polypeptides, assure leur repliement et leur localisation correctes. La SUMOylation de RanGAP1 est indispensable à son association avec la nucléoporine RanBP2 au niveau des filaments cytoplasmiques du complexe du pore nucléaire. Les résultats d’une analyse protéomique des composants de cette structure chez les mammifères ont révélé l’importance des complexes enrichis en protéines SUMO et leurs enzymes de conjugaison associées aux filaments du complexe du pore nucléaire.
Références : Matunis MJ, Blobel G, Coutavas E A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex (1996)
Mahajan R, Gerace L, Matunis MJ A small ubiquitin-related polypeptide involved in targeting RanGAP1 to the nuclear pore complex protein RanBP2 (1997)
Matunis MJ, Wu J, Blobel G SUMO-1 modification and its role in targeting the Ran GTPase-activating protein, RanGAP1, to the nuclear pore complex (1998)
Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ Proteomic analysis of the mammalian nuclear pore complex (2002)
Werner, A., Flotho, A., & Matunis, M. J The RanBP2/RanGAP1*SUMO1/Ubc9 SUMO E3 ligase is a multi-subunit autonomous disassembly machine (2012)
La sumoylation régule la fonction de très nombreuses protéines (des centaines et peut-être des milliers) par l’addition par une liaison iso-peptidiquecovalent entre l’extrémité C-terminale de SUMO et un résidu lysine de la protéine cible. Elle fait intervenir une cascade séquentielle de trois enzymes : E1 (complexe hétérodimérique d’activation), E2 (conjugaison), E3 (ligase). Les protéines-cibles possèdent le motif SIM (Sumo-Interacting Motif). La régulation se fait souvent par l’association de protéines cellulaires à la protéine modifiée. Cette association résulte de l’interaction non covalente entre un motif SIM (Sumo-Interacting Motif) sur la protéine-cible et une protéine modifiée. L’interaction implique, d’une part une courte séquence de 4 résidus aliphatiques (motif SIM) et une fente hydrophobe sur SUMO et, d’autre part des liaisons électrostatiques entre des acides aminés chargés positivement de SUMO et les résidus chargés négativement d’une séquence flanquant la portion hydrophobe du motif SLIM.
Article de synthèse : Yau TY, Sander W, Eidson C, Courey A. A SUMO Interacting Motifs: Structure and Function (2021)
Par son caractère réversible, lasumoylation confère aux protéines modifiées la capacité d’agir comme desinterrupteurs moléculaires et de réguler la biogenèse des ribosomes (maturation de l’ARNr par la ligase SUMO USP36 (Ubiquitin Specific Peptidase 36) et les protéines Nop58, Nop56, Nhp2 et DKC1), la maintenance de la structure du nucléole ou le cycle cellulaire (association de RanGAP1 modifié à l’appareil du fuseau mitotique). Dans le cytosol, après avoir lié SUMO-1, RanGAP1 se fixe sur les fibres cytoplasmiques du complexe du pore nucléaire en se liant à la volumineuse ( 358 kDa)nucléoporine RanBP2 (Ran GTP Binding Protein 2). Le complexe RanGAP1-RanBP2 régule l’import-export nucléaire du cargo (ARN, protéines). Dans le cytosol, la conversion de Ran-GTP en Ran-GDP, provoque la dissociation des complexes d’exportation nucléaire et la libération du cargo à l’extérieur du noyau.
La voie de la sumoylation des protéines est régulée : en plus des ligases qui ajoutent des protéines SUMO – ou des chaînes de poly-SUMO – sur les protéines cibles, il existe des protéases qui les retirent. En collaboration avec Michael N. Boddy (Department of Molecular and Cellular Biology, Scripps Research), Guy Salvesen et coll. (Sanford Burnham Prebys Medical Discovery Institute) ont étudié les enzymes de déSUMOylation. Les protéases SENP (Sentrin-specific Proteases) (SENP3, SENP5) sont des cystéine protéases qui coupent les liaisons isopeptidiques pour détacher les protéines SUMO de leurs protéines cibles. Cette étape de déconjugaison est essentielle pour l’assemblage et la maturation des sous-unités ribosomiques. Salvesen et coll. ont décrypté le mécanisme par lequel les ciseaux moléculaires SENP régulent la réversibilité du système SUMO en découpant les chaînes SUMO de manière aléatoire – stochastique – permettant une homéostasie rapide des protéines modifiées. Leurs études de la cinétique et de la spécificité des protéases SENP ont élucidé comment elles sélectionnent leurs substrats (SUMO-1, -2, -3) et catalysent la maturation des précurseurs (activité endopeptidase), ou la déconjugaison (activité isopeptidase).
Références : Mikolajczyk, J., Drag, M., Bekes, M., Cao, J. T., Ronai, Z., & Salvesen, G. S. Small Ubiquitin-related Modifier (SUMO)-specific Proteases: Profiling the specificities and activities of human SENPs (2007)
Drag M, Salvesen GS DeSUMOylating enzymes–SENPs (2008)
Kolli N, Mikolajczyk J, Drag M, Mukhopadhyay D, Moffatt N, Dasso M, Salvesen G, Wilkinson KD Distribution and paralogue specificity of mammalian deSUMOylating enzymes (2010)
Bekes M, Prudden J, Srikumar T, Raught B, Boddy MN, Salvesen GS The dynamics and mechanism of SUMO chain deconjugation by SUMO-specific proteases (2011)
Albrow VE, Ponder EL, Fasci D, Békés M, Deu E, Salvesen GS Development of small molecule inhibitors and probes of human SUMO deconjugating proteases (2011)
La biogenèse des ribosomes est un processus étroitement contrôlé. La nucléophosmine (NPM1/B23) (37 kDa), abondante dans le nucléole, est impliquée dans l’assemblage correct des sous-unités ribosomales. Agissant comme une chaperone, elle empêche l’agrégation des protéines ribosomales entre elles et facilite leur interaction avec l’ARNr. La nucléostémine, identifiée par Robert Tsai et Ronald McKay (National Institute of Neurological Disorders and Stroke) est une GTPase. Elle contrôle la prolifération cellulaire en interagissant avec des protéines comme p53 ou le régulateur du cycle cellulaire MDM2 (Murine Double Minute 2). Au cours du cycle cellulaire, l’activité de la nucléoplasmine et sa capacité à se lier aux acides nucléiques sont modulées par phosphorylation. Elle régule l’accès de la chromatine et le transport nucléocytoplasmique. Elle participe à la transcription, au clivage et à l’assemblage des pré-ribosomes au sein du nucléole.
Le nucléole intervient dans la régulation du cycle cellulaire par sumoylation du complexe RanGAP1-RanBP2 ; après fragmentation de l’enveloppe nucléaire, l’interaction entre les microtubules du fuseau mitotique et les kinétochores des chromosomes est sous le contrôle de RanGAP1-RanBP2 modifiéassurant ainsi la ségrégation correcte des chromosomes. La sumoylation agit comme un signal de rétention et de séquestration dans le nucléole de suppresseurs de tumeur (p53), bloquant ainsi l’interaction avec leurs cibles cytoplasmiques ou nucléaires.
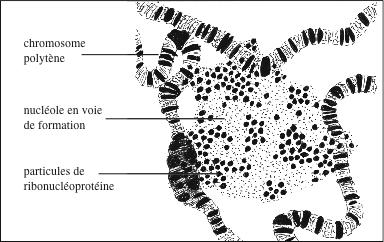 |
| Formation d’un nucléole in vitro. Un transgène de pré-ARNr a été artificiellement inséré dans un chromosome polytène ; le chromosome recombiné est placé dans un milieu contenant les ingrédients nécessaires à la transcription. Une matrice de protéines et d’ARN s’organise spontanément, au sein de laquelle apparaissent des ribonuléoprotéines 80S, les précurseurs des sous-unités ribosomiales. |
Chimie du noyau
Les chimistes commencèrent à s’intéresser au noyau au XIXe siècle, à une époque où la chimie organique structurale connaissait en Allemagne un spectaculaire développement. Entre les années 1840 et 1870, l’enseignement et la recherche en chimie physiologique se faisaient dans les écoles de médecine des universités allemandes, puis dans les facultés de philosophie (facultés des sciences). En 1845, les autorités de la Eberhard Karls Universität de Tübingen créèrent la première chaire de chimie physiologique et la confièrent à un pionnier de cette discipline : Julius E. Schlossberger. Au départ, cette chaire avait été créée « à titre personnel » ; elle devint permanente et le chimiste organicien Ernst Felix Immanuel Hoppe, qui se faisait appeler Hoppe-Seyler, prit la succession de Schlossberger. Hoppe-Seyler choisit comme thème de recherches la composition chimique du protoplasme.
Johan Friedrich Miescher a étudié la médecine à l’Université de Bâle. Atteint de surdité à la suite d’une attaque de typhus, il renonce à la pratique médicale, opte pour la recherche et suit une formation de chimiste organicien dans le laboratoire d’Adolf Stecker, à Göttingen. Il est convaincu que la composition chimique du noyau doit être radicalement différente de celle des autres parties de la cellule. A Tübingen, devenu l’élève d’Hoppe-Seyler, et malgré la mise en garde de son mentor sur la difficulté du projet, Miescher entreprend l’étude du noyau des leucocytes, un matériel expérimental doté de volumineux noyaux. Il en récolte de grandes quantités en lavant avec une solution de sulfate de sodium des pansements imprégnés de pus ; les protéines cellulaires sont éliminées par un traitement à la pepsine (un extrait d’estomac de porc) en milieu chlorhydrique ; le matériel est soumis à une longue hydrolyse acide diluée, puis à une extraction à l’éther pour éliminer les lipides. Une couche de matériel à l’interface eau/éther se sépare d’un culot insoluble dans l’eau et les solvants acides mais soluble en milieu alcalin en donnant un liquide visqueux ; par acidification progressive, Miescher précipite un matériel biologique d’acidité élevé (le plus acide jamais isolé) et riche en phosphore (10% de la masse). On ne connaissait à l’époque que deux composés biologiques contenant du phosphore : la caséine, une phosphoprotéine du lait isolée par Gerrit Mulder en 1837, et la lécithine, un phospholipide isolé dans le laboratoire d’Hoppe-Seyler. Convaincu d’avoir isolé une substance spécifique du noyau, Miesscher lui donne le nom de « nucléine » (1869) et rédige un article qu’il soumet pour publication à Hoppe-Seyler, éditeur de la revue Medizinisch-chemishe Untersuchungen. Confronté au caractère novateur, pour ne pas dire révolutionnaire, des résultats de Miescher, Hoppe-Seyler adopte une attitude prudente non dépourvue d’ambiguïté : il retarde pendant deux ans la publication du manuscrit et demande à deux de ses élèves de reproduire les résultats de Miescher en appliquant son protocole expérimental sur du sang (Pal Plosz) et de la caséine (Nicolai N. Lubavin). L’article est finalement publié en 1871. Miescher quitte le Schlosslaboratorium d’Hoppe-Seyler pour rejoindre Carl Ludwig, le réputé physiologiste de l’Université de Leipzig, puis occupe la chaire de physiologie de l’Université de Bâle, où il entreprend des travaux sur le sperme de saumon du Rhin, une source particulièrement riche en nucléine ; en 1868, il en isole un matériel basique riche en azote : la protamine, dont la nature protéique sera établie en 1894 par Albrecht Kossel. Plus tard, il démontre la liaison entre la nucléine acide et la protamine (histone) basique. Miescher était convaincu que la nucléine jouait un rôle dans la fécondation et l’hérédité. Il avait voulu en faire mention dans l’article de 1871, mais Hoppe-Seyler s’y était opposé. Miescher revint sur cette interprétation prémonitoire du rôle de la nucléine dans un article publié en 1874.
Références : Miescher F Über die chemische Zusammensetzung der Eiterzellen (1871)
Miescher F Über die Spermatozoen einiger Wirbeltiere (1874)
Le laboratoire de Felix Hoppe-Seyler (Université de Strasbourg) avait comme thème de recherche la composition chimique des tissus. En 1878, son assistant, K.L.M.L. Albrecht Kossel, isola l’hypoxanthine et la xanthine, après hydrolyse d’une préparation de nucléine de levure et de globules rouges. Ces deux bases n’étaient pas des inconnues ; la xanthine avait déjà été isolée par le chimiste Charles Frédéric Kuhlman et les deux purines, par Jean Piccard (École polytechnique fédérale, Zurich), à partir de nucléine de sperme de poisson. Kossel montra que xanthine et hypoxanthine proviennent bien de la nucléine et non des protéines, comme le soutenaient Georg Salomon et Russel H. Chittenden (Yale University). Ces deux bases puriques sont des produits de désamination formés par dégradation thermique de l’adénine et de la guanine. L’adénine fut isolée par Kossel, en 1885, à partir de tissu pancréatique et de nucléine. Richard Altman (Universität Leipzig) obtint une préparation de nucléine de levure très faiblement contaminée par des protéines ; en 1899, il proposa de remplacer le terme « nucléine » par « acide nucléique ». Dans l’hydrolysat de nucléine de levure d’Altman, Kossel caractérisa la présence d’une forte quantité d’acide phosphorique, de guanine, d’adénine et d’un produit ayant les propriétés d’un hydrate de carbone (1891). En 1893, Kossel et son étudiant Albert Neumann découvrirent les bases pyrimidiques thymine et cytosine. En 1901, un autre de ses étudiants, Alberto Ascoli, découvrit l’uracile à partir d’acide nucléique de levure. Après la découverte des bases entrant dans la composition des acides nucléiques – adénine, guanine, thymine, cytosine, uracile – Kossel abandonna progressivement leur étude au profit de celle des protéines ; il concevait difficilement que les acides nucléiques, composés de seulement cinq bases différentes, puissent rendre compte de la diversité du vivant ; les protéines, avec leurs vingt acides aminés différents, paraissaient de meilleures candidates. En 1884, Kossel découvrit l’histone, une protéine basique associée à l’ADN et différente de la protamine isolée par Friedrich Miescher. En 1895, il isola un nouvel acide aminé, l’histidine. Il reçut en 1910 le prix Nobel de médecine ou physiologie.
Références : Kuhlman CF Recherches scientifiques (1877)
Salomon G Ueber die Verbreitung und Entstehung von Hypoxanthin und Milchsaure im thierischen Organismen (1878)
Chittenden RH On the formation of hypoxanthine from albumin (1879)
Altman R Ueber Nucleinsauren (1889)
Kossel A, Neumann A Ueber das Thymin ein Spaltungsprodukt der Nucleinsaure (1893)
Ascoli A Ueber ein neues Spaltungsprodukt des Hefenucleins (1901)
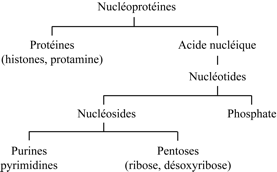 |
| Protocole d’hydrolyse chimique des nucléoprotéines. Ce schéma d’hydrolyse fut utilisé jusqu’au milieu du XXe siècle. L’hydrolyse chimique ménagée, combinée à l’hydrolyse enzymatique, libère un mélange de bases, pentoses, pentoses phosphate, nucléosides, nucléotides et oligonucléotides. Le terme « protamine » désigne aujourd’hui des protéines basiques exprimées dans les spermatocytes et jouant un rôle analogue à celui des histones. |
« A dull repetitive molecule »
Dans un texte dont j’ai omis de noter la référence, un auteur qualifie ainsi l’ADN : « a dull repetitive molecule incapable of fulfilling any important role ». Rappelons quelques faits : (i) Miescher isole la nucléine dans années 1860 ; à la fin des années 1940, on ne sait toujours pas avec certitude quel est le rôle joué par l’ADN dans la transmission des caractères héréditaires ; (ii) en 1879, Walther Flemming désigne par le terme « chromatine » le matériel nucléaire présent dans le noyau et qui fixe fortement les colorants basiques ; sa nature (protéines, acides nucléiques) est inconnue. En 1881, Eduard Zacharias montre que la chromatine réagit comme la nucléine à des traitements acide ou alcalin ; selon lui les deux termes désignent le même matériel nucléaire. (iii) jusqu’aux années 1940, on distingue l’acide nucléique de levure (acide zymonucléique) présent dans les végétaux, de l’acide thymonucléique présent dans les tissus animaux (car isolé à partir de thymus de veau). Dans les années 1920, le biochimiste J.W. Robert Feulgen (Christian-Albrecht Universität, Kiel) et le médecin Heinrich Rossenbeck mettent au point une méthode de coloration spécifique de l’acide thymonucléique. La technique comporte deux étapes : (i) une hydrolyse ménagée – acide chlorhydrique à 60°C – de l’échantillon tissulaire ; dans ces conditions les liaisons N-osidiques entre bases puriques et pentoses sont rompues ; le groupe aldéhydique du désoxyribose est libéré mais pas celui du ribose dont le groupe hydroxyle sur le C2 bloque l’hydrolyse acide ; (ii) coloration au réactif de Schiff (fuchsine basique décolorée au dioxyde de soufre SO2). Au contact des groupements aldéhydes du désoxyribose le réactif prend une coloration pourpre/violette. Le cytoplasme et les protéines ne sont pas colorés par la réaction de Feulgen. En appliquant ce qu’ils appelait la « réaction nucléale » sur une grande variété de tissus et d’organismes (grenouilles, rats, protozoaires, spermatozoïdes, tissus végétaux, germes de blé et de seigle) Feulgen et Rossenbeck montrent que l’acide thymonucléique est présent exclusivement dans le noyau de toutes les cellules animales et végétales. La chromatine est composée d’acide thymonucléique.
Dans les années 1920, Walter Jones et sa collaboratrice M. E. Perkins (Johns Hopkins University) mirent au point un protocole de purification de l’ARN qui évite sa dégradation par les ribonucléases. Dans les années 1920 et 1930, le biochimiste Johan Erik Jorpes (Karolinska Institutet, Stockholm) parvint à extraire et à purifier de l’ARN à partir de pancréas de bœuf, mettant ainsi fin au dogme selon lequel l’acide ribonucléique de levure (ARN) n’est présent que dans les tissus végétaux. La coloration de coupes provenant du même échantillon tissulaire par le réactif de Feulgen (ADN) ou par des colorants basiques (ADN et ARN) permet de mettre en évidence la présence des deux types d’acides nucléiques dans les cellules animales et végétales.
Références : Feulgen R, Rossenbeck H Mikroskopisch-chemischer Nachweis einer Nucleinsäure vom Typus der Thymonucleinsaure und die darauf beruhende elective Farbung vom Zelikernen in mikroskopischen Praparaten (1924)
Jones W, Perkins ME The Nucleotides of Yeast Nucleic Acid (1923)
Jones W, Perkins ME The Plant Nucleic Acid (1923)
Jorpes JE Zur Kenntnis der « Pankreasnucleinsäure » (1924)
La recherche sur les acides nucléiques, abandonnée par Kossel, fut poursuivie par ses élèves Walter Jones et Phoebus Levene. Il fallait d’abord connaître la formule chimique des bases qui avaient été isolées. En 1881, Emil Hermann Fischer eut recours aux techniques de la chimie organique structurale par analyse et rétro-synthèse mises au point par Friedrich Whöler pour établir, en 1828, la structure de l’urée. Fisher élucida la structure d’une série de composés apparentés (acide urique, xanthine, caféine, théobromine) et montra qu’ils dérivent d’une base azotée qu’il nomma purine par référence à sa parenté structurale avec l’urée. La structure moléculaire de l’adénine et de la guanine dérive de celle de la purine ; celle de la thymine et de la cytosine dérivent de la structure de la pyrimidine. Fisher et Julius Tafel apportèrent une contribution décisive à l’établissement de la structure des sucres, hexoses et pentoses. Je rappelle que c’est Fischer (prix Nobel de chimie en 1902) qui a découvert la liaison peptidique et montré que les protéines sont des polymères d’acides aminés (le terme « protéine » fut inventé par le chimiste Jön Jacob Berzelius et son emploi, généralisé à partir de 1839 par Gerardus Johannes Mulder, spécialiste des albumines).
Au moment où Phoebus A.T. Levene commence à s’intéresser aux acides nucléiques, Albrecht Kossel et ses associés en ont caractérisé les « building blocks » : les bases puriques – adénine, guanine – et pyrimidiques – thymine, cytosine, uracile – et un sucre, peut-être un pentose. Ils ont calculé que les acides nucléiques renferment autant de bases puriques que de bases pyrimidiques. Je rappelle qu’à cette époque, en plus des « acides nucléiques simples », comme l’acide inosinique (base – sucre – phosphate), isolé en 1847 par Justus Liebig dans un filtrat de muscle de bœuf, on connaît deux « acides nucléiques complexes » : l’acide zymonucléique et l’acide thymonucléique. En 1902, deux spécialistes de la chimie des protéines, Thomas B. Osborne et Isaac F. Harris (Connecticut Agricultural Experiment Station), proposent un modèle de structure pour l’acide zymonucléique de germe de blé, dans lequel le sucre et les bases sont attachés par des liaisons covalentes à un squelette de quatre phosphates, liés entre eux par des liaisons pyrophosphate. Le rapport entre les bases est : guanine/adénine/2 uracile, Osborne et Harris n’ayant pas détecté la cytosine.
Références : Osborne TB, Campbell GF The Nucleic Acid of the Embryo of Wheat and Its Protein Compounds (1900)
Osborne TB, Harris IF The Nucleic Acid of the Embryo of Wheat (1902)
Osborne TB, Harris IF Die nucleinsäure des weizenembryos (1902)
C’est Levene et ses collaborateurs qui découvrent comment les « pièces détachées » s’associent entre elles pour former des nucléosides, des nucléotides et des polynucléotides. Après ses études de médecine à Saint-Pétersbourg, Levene émigre aux États-Unis où il combine la pratique médicale dans le quartier déshérité du Lower East Side, à New York, avec un poste d’étudiant chercheur à Columbia University. Atteint de tuberculose, il est astreint à des périodes de convalescence, entre 1896 et 1905, qu’il met à profit pour accroître ses connaissances en chimie organique auprès d’Albrecht Kossel et d’Emil Fischer. En 1905, après avoir obtenu une position permanente au Rockefeller Institute for Medical Research, il crée un laboratoire de chimie organique qui au fil des publications (700 articles scientifiques) acquiert une réputation mondiale. Levene procède avec méthode : il commence par mettre au point les conditions expérimentales pour obtenir des produits d’hydrolyse des acides nucléiques à un degré de pureté permettant leur quantification. L’hydrolyse enzymatique a été utilisée par Walter J. Jones (John Hopkins University, avec des extraits de pancréas, riches en nucléases ; Levene et Walter Jacobs ont recours aux enzymes digestifs contenus dans les sécrétions d’une fistule intestinale. En 1923, Levene reçoit la visite d’ Ivan P. Pavlov, son ancien professeur de physiologie à l’Université de Saint-Petersbourg, prix Nobel de médecine ou physiologie 1904, de passage à New York. Pavlov avait mené des expériences impliquant le recueil par une fistule de la sécrétion gastrique d’un chien. Au cours d’un séjour à Saint-Pétersbourg, Levene essaie, sans grand succès, le contact in vitro d’acides nucléiques avec le suc gastrique. Il est alors décidé qu’ Efim S. London, un membre du laboratoire de Pavlov, fera un séjour au Rockefeller Institute ; en 1928, il introduit les acides nucléiques par une fistule dans l’estomac d’un chien et recueille les produits d’hydrolyse par une fistule intestinale.
Levene et J. A. Mandel montrent que l’acide thymonucléique est constitué de modules constitués d’une base – un sucre – un phosphate associés linéairement. La liaison entre la base et le sucre pourrait être de nature osidique. Plusieurs modules (nucléotides) pourraient s’associer par leurs phosphates pour constituer des polynucléotides. Les mêmes structures modulaires existent dans l’acide zymonucléique, comme le montrent Levene et Walter A. Jacobs ; ils isolent la guanosine à partir d’acide guanylique de levure ; ils identifient le sucre au désoxypentose (d-ribose), un glucide qui avait été synthétisé en 1891 par Emil Fischer et Oskar Piloty (Université Humboldt, Berlin). Levene crée les termes « nucléotide », « polynucléotide », « nucléoside ». Levene et Jacobs montrent que l’hydrolyse alcaline de l’acide inosinique libère une base et du ribose phosphate ; l’hydrolyse acide libère un nucléoside et du phosphate ; ces résultats plaident en faveur de la liaison des groupes phosphate aux bases par l’intermédiaire des sucres, l’ordre d’association étant : base – pentose – phosphate. Il existe une liaison β-N-glycosidique entre la base et le carbone C1’ du 2’-désoxyribose. Levene et London isolent à partir d’acide thymonucléique un guanine-désoxypentoside. Le sucre présent dans ce qu’on nommera plus tard ADN n’est pas un hexose, comme le prétendait Walter Jones, un élève d’Albrecht Kossel, mais un pentose, le désoxyribose, dont la structure 2-désoxyribo furanosique sera déterminée en 1935.
Références : Levene PA On the preparation of nucleic acids (1900)
Levene PA Hydrolysis of spleen-nucleic acid by dilute mineral acid (1904)
Walter J The action of boiled pancreas extract on yeast nucleic acid (1920)
Levene PA, Mandel JA Über die konstitution der thymo-nucleinsäure (1908)
Levene PA, Jacobs WA Über die hefe-nucleinsäure (1909)
Fischer E, Piloty O Ueber eine neue Pentonsäure und die zweite inactive Tryoxyglutarsäure (1891)
Levene PA, London ES Guaninedesoxypentoside from thymus nucleic acid (1929)
Levene PA, London ES The structure of thymonucleic acid (1929)
Levene publie en 1909 un schéma de structure des acides nucléiques comportant un anneau tétra-pyrophosphate ; les bases, attachées au C1′ du ribose, sont dirigées vers l’extérieur. Le rapport des bases est A/T/G/C/ pour l’ADN et A/G/C/U pour l’ARN (Walter Jones, 1914). En 1935, Levene et R. Stuart Tipson établissent la structure de l’acide nucléique (correcte pour l’ADN et presque correcte pour l’ARN. La digestion avec des ribonucléases permettra d’établir que, dans l’ARN, la liaison phosphodiester s’établit entre les C3′ et C5′ du pentose. En 1952, D.M. Brown et Alexander R. Todd (Cavendish Laboratory, University of Cambridge) montrent que dans les acides nucléiques les pentoses de deux nucléotides adjacents sont unis entre eux par des liaisons 3’-5’ phosphodiester. Le talon d’Achille de Levene reste le modèle de structure publié en 1909 sous le titre « Yeast nucleic acid ». En limitant l’ADN à un tétranucléotide, Levene est passé à côté de sa nature macromoléculaire. Après avoir eu connaissance des données expérimentales de Rudolf Signer (Universität Bern) et des chercheurs scandinaves Torbjörn Caspersson (Karolinska Institutet, Stockholm), Ivar Christian Bang et Olof Hammarsten (Uppsala) prouvant que l’on avait grandement sous-estimé la taille de l’ADN, Levene proposa le modèle du « polytétranucléotide » long d’une dizaine de nucléotides :
TACG TACG TACG TACG…
Ce modèle fut pendant quarante ans universellement adopté par la communauté scientifique Le généticien Max L.H. Delbrück (prix Nobel de médecine ou physiologie 1969 et père fondateur du Groupe du phage) a qualifié l’ADN de « molécule stupide » ce qui eut pour conséquence d’inciter les chercheurs à poursuivre la piste des protéines basiques associées à l’ADN. Albrecht Kossel a reçu le prix Nobel de physiologie ou médecine 1910 pour avoir élucidé la structure chimique des constituants des acides nucléiques. Le jury Nobel s’est montré très généreux envers Alexander Todd en lui attribuant le prix Nobel de chimie 1957 « for his work on nucleotides and nucleotide co-enzymes » ; par contre, il n’a retenu du travail de Levene que le « catastrophique » modèle du tétranucléotide, faisant abstraction de son apport considérable à la connaissance de la structure des acides nucléiques.
Références : Bang I Untersuchungen über die Guanylsäure (1910)
Brown DM, Todd AR Nucleotides. Part X. Some observations on the structure and chemical behaviour of the nucleic acids
Brown DM, Heppel LA, Hilmoe RJ Nucleotides. Part XXIV. The action of some nucleases on simple esters of mononucleotides (1954)
Hammarsten O Zur Kentnis der Nukleoproteide (1894)
Jones W Nucleic Acids, Their Chemical Properties and Physiological Conduct (1914)
Levene PA Yeast nucleic acid (1909)
Levene PA, Jacobs WA Über die Pentose in den Nucleinsaüren (1909)
Levene PA, Jacobs WA Über hefe-nucleinsäure (1909)
Levene PA On the biochemistry of nucleic acids (1910)
Levene PA, Tipson RS The ring structure of thymidine (1935)
Signer R, Caspersson TO, Hammarsten E Molecular Shape and Size of Thymonucleic Acid (1938)
Whitfeld PR, Markham R The natural configuration of the purine nucleotides in ribonucleic acids: chemical hydrolysis of the dinucleoside phosphates (1953)
Note : Pour la rédaction de ce sous-chapitre, j’ai consulté les articles originaux chaque fois que cela m’a été possible. J’ai, en outre, puisé des informations dans l’excellente revue d’Eugenio Rixione et Lourdes Ruiz-Zamarripa : « The “scientific catastrophe” in nucleic acids research that boosted molecular biology » (2019).
La situation évolua rapidement lorsqu’un certain nombre de groupes décidèrent de perfectionner la méthode d’extraction de l’ADN. John M. Gulland et Denis O. Jordan (University College, Nottingham) évitèrent le recours à l’hydrolyse chimique acide ou alcaline et conservèrent la préparation à un pH voisin de la neutralité à toutes les étapes de la purification ; ils isolèrent à partir de thymus de veau des fibres d’ADN peu dégradées. Avec ce produit très purifié, ils démontrèrent en 1947 l’existence de liaisons hydrogène entre les groupes aminés et hydroxyle des bases par titration électrométrique avec un acide fort ou une base forte. C’était un premier pas vers la structure hélicoïdale de l’ADN.
Les chercheurs scandinaves : Olof Hammarsten (Uppsala universitet), Einar Hammarsten, Harald Hammarsten, Ivar Bang (Lund universitet), Theodor Svedberg, Torbjörn O. Caspersson (Karolinska Institutet) adoptèrent une stratégie similaire : extraction de l’ADN à basse température et à un pH voisin de la neutralité. Ils séparèrent un matériel blanc, floconneux, très visqueux, précipitant spontanément en longues fibres au fond des récipients. Ivar Bang ajouta au processus de purification de l’ADN une étape de précipitation par un sel de sodium, pour diminuer au maximum la contamination par les protéines (soupçonnées par certains d’être le support de l’hérédité). Son élève, Einar Hammarsten, fit une évaluation de la taille de ce qu’il croyait être des « particules colloïdales » d’ADN, selon la conception en vigueur à l’époque. Cette notion de colloïde avait été introduite par le physico-chimiste Thomas Graham alors qu’il s’intéressait à la nature physique des protéines. Il tenta de séparer par dialyse d’extraits organiques des cristaux et des « colloïdes », et parvint à la conclusion que certaines protéines, comme la gélatine, étaient des colloïdes alors que d’autres étaient cristallisables. Il lança l’idée que l’état colloïdal était une spécificité du vivant.
Pour mesure la taille des particules d’acide thymonucléique, Einar Hammarsten et Torbjörn Caspersson sollicitèrent, en 1936, la collaboration du biophysicien Rudolf Signer. Ancien collaborateur d’Hermann Staudinger (prix Nobel de chimie en 1953, créateur, en 1922, du terme « macromolécule »), Signer était un spécialiste de la biréfringence de flux. Cette technique met en jeu la double réfraction de rayons lumineux et permet d’estimer la taille et la forme de molécules en solution. Lorsque l’on force la solution à s’écouler dans un tube de très faible diamètre toutes les molécules ont tendance à s’orienter dans le même sens, qui est le sens du flux. Dès 1934, Torbjörn Caspersson et Einar Hammarsten avaient démontré que l’ADN est une macromolécule. Les résultats définitifs publiés en 1938 avec Rudolf Signer créèrent la surprise, pour ne pas dire le choc : la longueur de la molécule d’ADN équivalait à trois cent fois son diamètre et sa masse était comprise entre 500.000 et 1.000.000 ! La technique permettait même de conclure que le plan des bases devait être perpendiculaire au grand axe de la molécule ! En 1950, Signer analysa une préparation dont les molécules avaient une masse de 7.000.000 ! Les Scandinaves et leurs associés devinrent les grands pourvoyeurs d’ADN purifié. En 1937, Torbjörn Caspersson donna l’échantillon d’ADN de thymus de veau qui permit au physicien William Astbury (Davy-Faraday Laboratory, Royal Institution, London) d’entreprendre son travail de pionnier sur la structure spatiale de cette macromolécule par diffraction des rayons X. Rudolf Signer fournit les préparations à partir desquelles les physiciens britanniques établirent la structure en double hélice de l’ADN.
Devant ces résultats, Phoebus Levene jugea qu’il devait réévaluer la taille des molécules d’ADN qu’il utilisait ; avec Gerhard Schmidt, il mesura leur masse par centrifugation à haute vitesse. Les valeurs mesurées étaient comprises entre 200.000 et 1.000.000. Levene ne voulut pas voir à quel point ce résultat invalidait son modèle du poly-tétranucléotide qui, en s’imposant à la communauté scientifique, avait freiné tout progrès des connaissances sur la fonction des acides nucléiques dans la cellule.
Division cellulaire, mitose, méïose
En 1824, Jean-Louis Prévost, qui deviendra le premier titulaire de la chaire de physiologie de la Faculté de Médecine de l’Université de Genève, et l’apprenti pharmacien Jean-Baptiste Dumas, qui allait devenir un chimiste de renom international, mettent en évidence le rôle des « produits du testicule et de l’ovaire » (spermatozoïdes et ovules) dans la fécondation, la segmentation de l’œuf fécondé et les premiers stades du développement de l’embryon. La segmentation de l’œuf en 2, 4, 8… cellules embryonnaires (blastomères) et la formation de la morula (embryon au stade précoce du développement) fut confirmée par Mauro Rusconi (Université de Paris) en 1826. Karl E. R. von Baër, professeur d’Anatomie, puis de Zoologie à l’Université de Königsberg et pionnier de l’embryologie découvre, en 1827, l’ovule des mammifères et déclare qu’il est à l’origine du fœtus.
Robert Remak, un élève de Johannes Müller, énonce, en 1852, que toute cellule provient de la division d’une cellule préexistante, que la division du noyau (caryocinèse) précède la division cellulaire (cytocinèse) ; ses travaux sur l’œuf fécondé, à l’hôpital de la Charité, à Berlin, mirent en évidence le rôle des trois feuillets embryonnaires, qu’il nomma « ectoderme, mésoderme et endoderme » ; c’est à partir de ces couches de cellules que se fait la différentiation et le développement des organes de l’embryon. En 1858, il nota l’apparition de filaments dans le noyau au début de la division cellulaire, une observation qui avait été faite dix ans plus tôt par Wilhelm F.B. Hofmeister dans le noyau des cellules végétales.
Les premières études sur la la fécondation chez des organismes vivants furent réalisées par Oscar Hertwig, professeur d’Anatomie à l’Université de Berlin et disciple du botaniste Ernst Haeckel, professeur à l’Université d’Iéna, directeur de l’Institut de Zoologie et propagandiste du darwinisme en Allemagne. Haeckel avait la conviction qu’il existait une relation entre hérédité et noyau. En 1850, Nicholas A. Warneck avait signalé que l’œuf fécondé renferme deux pronuclei (le terme « pronucléus » désigne le noyau de l’ovule ou du spermatozoïde, après fécondation). En 1873, le zoologiste Johann A.O. Bütschli fit une observation similaire : l’œuf fécondé d’un Nématode possède deux noyaux et ces noyaux fusionnent, phénomène qui fut aussi remarqué par L. Auerbach en 1874.
Hertwig séjourna à la station de zoologie marine de Villefranche-sur-Mer à la recherche d’un matériel expérimental adapté à ses travaux ; il porta son choix sur l’étoile de mer et surtout sur l’œuf d’oursin, dont la transparence lui permit d’examiner l’intérieur de la cellule au microscope et de découvrir, en 1876, le mécanisme de la fertilisation. Le phénomène de fusion des noyaux des cellules reproductrices mâle et femelle décrit par Hertwig fit l’objet d’études approfondies par Hermann Fol (Université de Genève), un autre élève d’Ernst Haeckel. Après fécondation, le noyau provenant du spermatozoïde est d’abord situé à la périphérie de l’ovule, puis il s’en rapproche, fusionne avec lui et donne naissance à une cellule-œuf. Hertwig rejetait l’idée que le cytoplasme puisse être le gardien de l’hérédité en se basant sur un raisonnement qui sera souvent repris par la suite : si le cytoplasme était le site de transmission des caractères héréditaires, on s’attendrait à ce que le cytoplasme de l’ovule et celui du spermatozoïde aient à peu près le même volume, ce qui n’est pas le cas ; celui de l’ovule est beaucoup plus volumineux que celui du spermatozoïde. Par contre, les noyaux du spermatozoïde et de l’ovule ont sensiblement la même taille.
Références : Prévost JL, Dumas JB Nouvelle théorie de la génération (1824)
Rusconi M Développement de la grenouille commune depuis le moment de sa naissance jusqu’à son état parfait (1826)
von Baër KER De ovi mammalium et hominis genesi epistola (1827)
Remak R Untersuchungen über die Entwickelung der Wirbelthiere (1855)
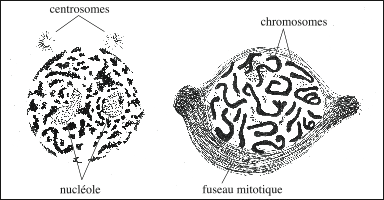 |
| Condensation de la chromatine en chromosomes pendant la transition de l’interphase à la prophase de la mitose. Dessin de gauche : enroulement des chromosomes en fibres ; dédoublement du centrosome. Dessin de droite : à la prophase, les chromatides appariées prennent l’apparence de filaments épais, les microtubules du fuseau mitotique sont visibles, les centrosomes sont diamétralement opposés, de part et d’autre du noyau. |
Les chromosomes ne sont pas visibles pendant l’interphase (entre deux divisions cellulaires) ; les seuls qui le soient sont les chromosomes géants (chromosomes polythènes). Ils ont été découverts, en 1881, chez les Chironomidae par Edouard-Gérard Balbiani, collaborateur de Claude Bernard puis titulaire de la chaire d’Embryologie comparée au Collège de France, et, en 1882, par Walther Flemming, professeur d’Anatomie à l’Université de Kiel, dans les glandes salivaires de Chironimus. Les chromosomes polythènes présentent des renflements (puffs) dénommés « anneaux de Balbiani », qui sont des sites de transcription active. Les chromosomes des cellules eucaryotes ne deviennent visibles qu’au moment de la mitose de la division cellulaire. La condensation de la chromatine en chromosomes fut décrite, en 1842, dans le noyau des cellules végétales par Karl Wilhelm von Nägeli, titulaire de la chaire de Botanique de l’Université de Zurich.
Balbiani entreprend l’étude de la reproduction sexuée sans parvenir à dégager une image claire et complète de la mitose, en partie à cause du matériel expérimental qu’il a choisi : les Protozoaires possèdent deux noyaux (macronucléus et micronucléus), ce qui complique singulièrement l’interprétation des résultats. Il utilise aussi les cellules de l’épithélium ovarien de la sauterelle Stenobothorus, sans parvenir à l’élucidation complète du processus ; il donne cependant une image assez exacte de la métaphase de la mitose chez les Protozoaires, et réalise une série d’observations pertinentes : (i) le noyau disparaît au début du processus mitotique ; (ii) des « bâtonnets » de tailles inégales apparaissent ; (iii) chaque bâtonnet donne naissance à deux bâtonnets. Leur existence avait déjà été signalée par Edouard van Beneden, professeur de zoologie et d’anatomie comparée à l’Université de Liège, ainsi que leur regroupement au voisinage de la plaque équatoriale et leur migration, en s’éloignant les uns des autres, vers les ébauches de noyaux des deux cellules filles.
La description des principales étapes de la division cellulaire par mitose chez les végétaux est l’oeuvre d’Eduard A. Strasburger, professeur de Botanique à la Jena Universität puis au Botanisches Institut, Universität Bonn. Il a inventé les termes « cytoplasme » et « nucléoplasme » (1882), qui décrivent plus exactement la réalité que « protoplasme ». Strasburger a concentré ses observations sur le comportement et le rôle du noyau : sa division pour donner le noyaux des cellules-fille ; son implication dans la transmission des caractères héréditaires au cours de la fertilisation des plantes. L’artisan de nos connaissances sur la mitose des cellules animales (nageoires et branchies de la salamandre) est Walther Flemming, professeur d’Anatomie (Christian-Albrechts-Universität zu Kiel). Il est l’inventeur des termes « chromatine » – la substance nucléaire acide qui fixe les colorants basiques comme l’aniline -, et « mitose », par référence à l’apparition de filaments lors de la division cellulaire. Les travaux sur la mitose étaient dans l’air du temps, comme on peut en juger par les notables contributions de J.A. Otto Bütschli (Christian-Albrechts-Universität zu Kiel), Waclaw Mayzel (Faculté de Médecine de l’École générale, Varsovie) ; W. Schleicher, l’inventeur du mot « caryocinèse » – la division du noyau (1879) ; Petr I. Peremeschko, professeur d’Histologie (Universität Kiev).
Mitose
| prophase | condensation de la chromatine en chromosomes |
| pro-métaphase | disposition en aster des chromosomes |
| métaphase | formation de la plaque équatoriale alignement des paires de chromatides dans le plan équatorial du fuseau mitotique |
| anaphase | disposition en double étoile des chromatides migration vers les pôles du fuseau mitotique |
| télophase | condensation des paquets de chromatides reconstruction du noyau |
Références : Balbiani, E.G Sur la structure du noyau des cellules salivaires chez les larves de Chironomus (1881)
Flemming W Zellsubstanz, Kern und Zelltheilung (1885)
Strasburger EA Über Zellbildung und Zelltheilung (1876)
Flemming W Kern und Zellteilung (1882)
Bütschli JAO Studien über die ersten Entwicklungsvorgänge der Eizelle, die Zelltheilung und die Conjugation der Infusorien (1876)
Mayzel W Ueber eigenthümliche Vorgänge bei der Theilung der Kerne in Epithelialzellen (1875)
Note : Les progrès des connaissances sur la division cellulaire ont été rendus possibles par l’amélioration de la qualité (aberrations) et du pouvoir de résolution des microscopes pendant la décade 1830-1839, et aux nouvelles méthodes de coloration de la chromatine. Eduard Strasburger était particulièrement réputé pour son expertise en ce domaine.
Strasburger était un disciple du botaniste Ernst Haeckel. Il étudiait la fécondation chez les conifères et les plantes à fleurs ; chez l’éphémère (Tradescantia) ou fritillaire (Fritillaria persica), il est possible de suivre au microscope le passage du noyau mâle dans le tube pollinique et sa progression vers le sac embryonnaire en traversant le pistil ; cette particularité permet de suivre la fusion des noyaux mâle et femelle. Strasburger réalisa que la division des cellules végétales et des cellules animales présente de grandes similitudes. En 1879, il décrivit les étapes de la mitose, la division de la cellule en deux cellules filles, la scission concomitante du noyau de la cellule mère en deux noyaux, l’apparition dans le noyau de bâtonnets fortement colorables et leur scission en deux. Strasburger avait ses limites ; il commit des erreurs d’appréciation. Comme Balbiani, il crut que la scission des chromosomes était transversale. Il reprit à son compte la thèse du cytoblastème exposée par Schleiden dans l’édition de 1875 de « Zellbildung und Zellteilung », et conclut que le noyau était synthétisé de novo. Il soutint que le mode de division différait selon le type de cellule. Comme auteur, Strasburger détient un record de longévité : son « Lehrbuch der Botanik für Hochschulen », publié en 1894, devint un « best seller » dont les éditions se succédèrent pendant tout le XXe siècle.
Walter Flemming utilisait comme matériel expérimental la salamandre (Salamandra maculata) dont certaines cellules possèdent de volumineux noyaux. Il observa la duplication et la scission longitudinale des « filaments » au cours de la mitose des cellules épithéliales de la larve. Fleming ne fut pas le premier à dire que la scission est longitudinale et non transversale comme le croyaient Balbiani et Strasburger ; W. Schleicher l’avait vu avant lui. Tout en contestant les résultats de Schleicher, Strasburger eut l’honnêteté de répéter ses expériences sur des plantes à fleur (ses premiers travaux portaient sur les conifères). Enfin convaincu que la scission était bien longitudinale, il obligea son éditeur à interrompre la vente de « Zellbildung und Zellteilung » et racheta, sur ses deniers, les exemplaires déjà vendus. En 1878, Fleming donna une image d’ensemble de la mitose assez conforme à la réalité, à l’exception d’un fait capital qui lui échappa totalement : la migration vers les pôles opposés de la cellule des deux nouveaux filaments après la scission. Si Strasburger et Fleming mentionnèrent l’existence du fuseau mitotique, ils eurent du mal à interpréter correctement son rôle dans la division cellulaire. Ils ne furent pas les seuls ; le zoologiste Johan A.O. Büstchli et Oskar Hertwig croyaient que les filaments (chromosomes) étaient le produit de la dilatation des fibres du fuseau mitotique.
Ascaris megalocephala est un vers nématode qui parasite de l’intestin des chevaux. Comme matériel expérimental, il présente de nombreux avantages : (i) l’utérus développé de la femelle adulte se prête particulièrement bien à la dissection, mettant en évidence les organes sexuels ; (ii) les cellules somatiques deviennent transparentes après un traitement approprié à l’alcool ; (iii) les cellules ne possèdent qu’un nombre réduit de chromosomes (quatre chez Ascaris megalocephala de la sous-espèce univalens, deux dans d’autres espèces) ; (iv) les chromosomes sont bien individualisés et conservent leurs caractères morphologiques d’une génération à l’autre. C’est sur ce matériel biologique qu’Édouard J.L.M. van Beneden (Université de Liège), puis Theodor Boveri (Zoologisches Institut Munchen, puis Universität Würzburg) ont étudié la maturation et la fécondation de l’œuf, et découvert les étapes de la division cellulaire réductionnelle appelée « méïose ».
Sous l’égide de son père, Pierre-Joseph van Beneden, professeur de Zoologie, Anatomie comparée et Paléontologie à l’Université Catholique de Louvain, le jeune Édouard s’intéressa aux vers et à leurs embryons ; il découvrit ainsi la « cellule-œuf » d’Ascaris megalocephala. Disposant d’un excellent microscope fabriqué à Paris par Friedrich Edmund Hartnack – et Carl Zeiss pour l’optique -, il observa en détail la morphologie de l’œuf, la fécondation et le début du développement embryonnaire. L’Université Catholique de Louvain ayant écarté sa candidature à un poste de professeur de Physiologie, van Beneden occupa la chaire de Zoologie, Anatomie et Physiologie comparées, puis d’Embryologie à l’Université de Liège. Dans sa publication de 1883, Van Beneden décrivit la réduction de moitié du nombre de chromosomes dans le noyau des ovules et dans celui des spermatozoïdes, par rapport aux noyaux des cellules somatiques. Il commit cependant l’erreur de croire que la méiose est une division cellulaire d’un type particulier, avec expulsion d’un globule polaire. Boveri proposa un mécanisme de la division réductive en deux étapes, dont les détails furent précisés par Oscar Hertwig (Universität Berlin).
Méïose
| méïose I | réplication de l’ADN de la cellule-mère (2n chromosomes) condensation de la chromatine en chromosomes première division du noyau |
| cytocinèse | première division du cytoplasme = 2 cellules-fille (n chromosomes) |
| méïose II | condensation de la chromatine en chromosomes deuxième division du noyau |
| cytocinèse | deuxième division du cytoplasme = 4 cellules-fille (n chromosomes) |
La réplication de l’ADN chromosomique de la cellule-mère donne naissance à deux chromatides par chromosome, comme dans la mitose. Au cours de celle-ci, les chromatides-sœurs provenant de chaque chromosome homologue demeurent liées l’une à l’autre au niveau d’une structure nucléoprotéique appelée « kinétochore » jusqu’au moment de leur séparation et de leur migration vers les pôles opposés du fuseau mitotique. Dans la méiose, les paires de chromatides provenant de deux chromosomes homologues s’apparient, formant des groupes de quatre chromatides qui s’alignent dans le plan équatorial du fuseau mitotique. La cellule diploïde subit deux divisions successives (méiose I et méiose II) donnant naissance à quatre gamètes haploïdes, du moins pour les spermatozoïdes. Pour les gamètes femelles (ovules), la méïose I aboutit à la production d’un ovule et d’un globule polaire (corps polaire) ; il en va de même de la méïose II. La division d’un ovule diploïde mène in fine à la production d’un pronucléus haploïde et de deux ou trois globules polaires.
 |
| Mitose et méïose. Au début de la mitose, chaque chromosome d’une paire homologue est dupliqué en deux chromatides visibles au microscope optique à la fin de la prophase. Elles sont liées l’une à l’autre par leurs centromères au niveau du kinétochore. Au début de la méïose (stade diplotène), deux paires de chromatides, provenant d’une paire de chromosomes homologues, s’associent en des points particuliers – les chiasmas – pour former des tétrades, ou bivalents. Du matériel génétique est échangé entre chromosomes homologues (flèches). |
La contribution de Boveri ne se limita pas à la découverte du mécanisme de la méiose. Walther Flemming avait noté la présence de « filaments » dans la cellule en interphase, mais il avait commis l’erreur de conclure que ces filaments étaient jointifs. En 1887 et 1888, après un examen attentif du comportement des chromosomes pendant le développement embryonnaire de l’œuf (zygote) et les divisions des premières cellules (blastomères), Boveri arriva à la conclusion que le nombre et la morphologie des chromosomes restent inchangés au cours de la division du noyau. L’individualité des chromosomes est conservée à l’interphase, comme à la mitose (même si les chromosomes perdent leur forme condensée et deviennent moins visibles) et au cours des divisions successives de la cellule. C’était un argument décisif en faveur de leur rôle dans la transmission des caractères héréditaires. Boveri fut l’un des premiers à attribuer un caractère morphologique spécifique (un trait phénotypique) à un chromosome particulier ; il eut l’intuition que la variabilité et le nombre élevé de caractères héréditaires d’un individu pouvaient s’expliquer par des échanges de « facteurs particulaires » entre chromosomes. Par contre, il fut moins perspicace en interprétant la faible colorabilité des chromosomes à l’interphase comme révélant une perte de substance chromatique ; les chromosomes se rechargeaient en chromatine au début de la division cellulaire. Les cellules diploïdes possèdent deux copies de chaque chromosome dont l’une est fournie par le père (le mâle) et l’autre par la mère (la femelle). Se basant sur des critères essentiellement microscopiques, les généticiens regroupent les chromosomes par paires et leur attribuent un numéro. Il existe une paire de chromosomes sexuels identiques chez la femelle (XX), mais pas chez le mâle (XY). Les gamètes renferment une seule copie de chaque chromosome ; chez les animaux, ils portent le nom de sperme et d’œuf. Flemming publia en 1882 un ouvrage célèbre dans lequel il fit la synthèse des connaissances sur la mitose : « Substance cellulaire, noyau et division cellulaire ».
Références : Van Beneden E Recherches sur la maturation de l’œuf et la fécondation (1883)
Hertwig O Beitrage zur Kenntniss der Bildung, Befruchtung und Theilung des Thierischen Eies (1876)
Flemming W Beiträge zur Kenntniss der Zelle und ihrer Lebenserschemungen, Theil I (1879)
Flemming W Zellsubstanz, Kern und Zelltheilung (1882)
Terminologie
La partition du matériel nucléaire et des chromosomes entre les cellules filles fut décrite en 1878 par W. Schleicher sur des cellules de larves d’Amphibiens ou de cartilage crânien. Il créa le terme « caryocinèse ». Walter Flemming, en 1882, utilisa celui de « caryo-mitose », qui sera abrégé en « mitose » ; il créa aussi les termes « asters » (formations étoilées apparaissant aux extrémités du fuseau mitotique et à partir desquelles rayonnent les microtubules), « chromatine » (matériel nucléaire possédant une forte affinité pour les dérivés basiques de l’aniline). Les termes « prophase, métaphase, anaphase, cytoplasme, nucléoplasme, diploïde, haploïde, gamète » furent proposés par Eduard Strasburger, et « télophase », par l’anatomiste Martin Heidenhain, en 1894. Le botaniste John Bretland Farmer et J.E. Moore proposèrent le terme « méïose » vers 1905. Les termes « gène, génétique » furent créés par William Bateson, en 1902. Le terme « chromosome » fut utilisé pour la première fois en 1888 par l’anatomiste Heinrich von Waldeyer-Hartz à propos du matériel nucléaire condensé, colorable, apparaissant au début de la division cellulaire ; ce terme désigne aujourd’hui une longue molécule d’ADN bicaténaire associée à des protéines, en particulier à des polypeptides fortement basiques appelés histones.
Centrosome
Le centrosome est un organite subcellulaire unique, sans membrane, formé d’une paire de centrioles entourés d’un matériel péricentriolaire amorphe de nature protéique. Ce « centre organisateur des microtubules » exerce une triple fonction dans : (i) la division cellulaire (mitose et méïose) ; (ii) le transport intracellulaire ; la morphologie de la cellule animale (la cellule végétale est dépourvue de nucléole). Les microtubules sont des cylindres creux assemblant des dimères de tubuline, dont la nucléation et l’ancrage se fait à partir du matériel péri-centriolaire. Les microtubules sont l’un des trois composants du cytosquelette, avec les microfilaments d’actine et les filaments intermédiaires. Lors de la division cellulaire, leur polymérisation donne naissance au fuseau mitotique bipolaire aux extrémités duquel se trouve une « sphère d’attraction » (aster). Les chromosomes s’accrochent par leur kinétochore aux fibres du fuseau ; celui-ci assure l’alignement des chromosomes dans le plan équatorial, la migration des chromatides vers les pôles, et leur égale répartition entre les cellules-filles. Dans la cellule en interphase, les microtubules du centre organisateur servent de « rails » aux vésicules assurant le transport antérograde de protéines et de lipides du réticulum endoplasmique à la membrane plasmique, et le transport vésiculaire rétrograde de la membrane plasmique vers les compartiments membranaires internes. Les microtubules participent, avec les filaments d’actine et les filaments intermédiaires, à la constitution du cytosquelette, qui confère forme et polarité à la cellule et qui, dans certains cas, lui permet de se déplacer.
Vers la fin du XIXe siècle, la réalité de l’existence du centrosome fut matière à d’âpres débats ; pour certains, c’était un artefact de fixation. En 1873, alors qu’il étudiait la fertilisation chez les œufs d’oursins et chez l’étoile de mer Asteria glacialis, le zoologiste Hermann Fol nota la présence d’un petit corps distinct au centre des asters. Il déduisit de ses observations que ce « centre d’organisation » est l’organe dynamique de la division cellulaire. En pénétrant dans l’ovule, le spermatozoïde apporte son « centre de division », qui se divise pour former les deux pôles du fuseau achromatique. Le centrosome ne fut réellement admis dans la panoplie des structures subcellulaires qu’à la suite des observations de Walther Flemming ; en 1875, il décrivit la présence d’un petit corps dense au centre des asters qui apparaissent lors de la division des cellules de salamandre. Dans son ouvrage de 1882, il suggéra que ce petit corps est un organite permanent de la cellule et qu’il a un comportement dynamique ; il se divise en deux avant la mitose pour former les deux pôles du fuseau.
En 1875-1876, W.A. Oscar Hertwig a mené une série de travaux sur la fécondation de l’œuf d’oursin (Toxopneustes lividus) à l’issue desquels il a prouvé que la fécondation consiste en la fusion des noyaux (syngamie) du spermatozoïde et de l’ovule. Le pronoyau libéré par le spermatozoïde se déplace vers le noyau femelle guidé par le centrosome. En plus d’apporter le matériel génétique complémentaire contenu dans le noyau mâle, le spermatozoïde fournit le centre cinétique nécessaire à la première division de l’œuf. Hertwig montra aussi que l’aster se divise en deux pour former les pôles du fuseau mitotique.
Références : Fol H Sur les phénomènes intimes de la division cellulaire (1877)
Fol H Sur les phénomènes intimes de la fécondation (1877)
Flemming W Sur la connaissance de la cellule et de ses phénomènes de division (1878)
Flemming W Zellsubstanz, Kern und Zelltheilung (1882)
Hertwig WAO Beiträge zur Kenntniss der Bildung, Befruchtung und Theilung des thierischen Eies (1876)
Hertwig WAO Das Problem der Befruchtung und der Isotropie des Eies, eine Theorie der Vererbung (1884)
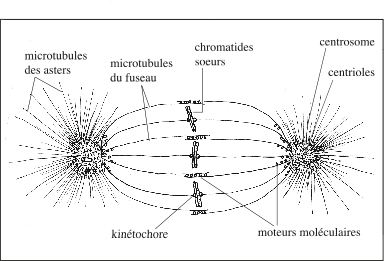 |
| Cellule en mitose. Pendant la phase S du cycle cellulaire, les deux centrioles se dédoublent. Au début de la division cellulaire, les deux nouveaux centrosomes s’éloignent l’un de l’autre pour former les pôles du fuseau mitotique. Dans le plan équatorial du fuseau, les paires de chromatides sont liées aux microtubules du fuseau par leurs kinétochores. Les chromatides se meuvent le long des microtubules du fuseau à l’aide de moteurs moléculaires (dynéines et kinésines) associés aux kinétochores. |
Une vive compétition a opposé Édouard J.L.M. Van Beneden (Université de Liège) et Theodor H. Boveri (Universität Würzburg) avec, à la clef, des contestations sur la priorité des découvertes du rôle du centrosome. En 1876, Van Beneden et Adolphe Neyt étudiaient les structures apparaissant au cours de la mitose de l’œuf ou des blastomères du nématode Ascaris megalocephala. En 1887, ils identifièrent à proximité du noyau, une « sphère d’attraction » qui persiste pendant l’interphase. Il la considéra comme un organite à part entière des cellules, même en dehors de la mitose. La sphère dérive par autoréplication d’un corpuscule préexistant ; sa division en deux corpuscules précède celle du noyau et l’apparition des « filaments » (chromosomes). Van Beneden attribua au « corpuscule central » un rôle essentiel dans la division cellulaire (karyocinèse et cytodiérèse). Des cellules, comme les neurones, qui ne se divisent pas, n’ont pas de centrosome fonctionnel.
Références : Van Beneden E Recherches sur la maturation de l’œuf, la fécondation et la division cellulaire (1883)
Van Beneden E, Neyt A Nouvelles recherches sur la fécondation et la division mitosique chez l’Ascaride mégalocéphale (1877)
En étudiant la fécondation et la division de l’œuf d’Ascaris megalocephala, Theodor Boveri observa la structure qui ne s’appelait pas encore « centrosome ». Il décrivit son comportement cyclique avant et pendant la prophase de la mitose et son dédoublement en se séparant au cours de l’anaphase et de la télophase. En 1888, l’observation des centrosomes de grande taille présents dans les œufs révéla la présence d’un granule dense que Boveri nomma « centriole » et de deux cylindres creux qu’il appela « centriole-mère » et « centriole-fille », disposés à angle droit l’un par rapport à l’autre. La paroi des centrioles est formée de microtubules arrangés en triplets, généralement au nombre de neuf. Certains protistes, les cellules végétales et des mycètes sont dépourvus de centrioles. En 1888, Theodor Boveri entreprit de faire une synthèse des données issues des travaux et propose le terme « centrosome » pour l’organite qu’il considéra comme une structure indépendante et permanente des cellules, transmise aux cellules-filles lors de la division cellulaire. Les asters et les centres de division ne sont pas des structures éphémères apparaissant à chaque mitose, comme on le croyait. Bovery fit du centrosome le « centre dynamique de la cellule, le centre de division » autour duquel s’organise la partition de l’œuf. Boveri a reconnu l’antériorité d’Hermann Fol dans la découverte des asters.
Références : Boveri T Sur la contribution du spermatozoïde à la division de l’ovule (1887)
Boveri T Zellen-Studien II: Die Befruchtung und Theilung des Eies von Ascaris megalocephala (1888)
Boveri T Sur la nature des centrosomes . Études cellulaires (1900)
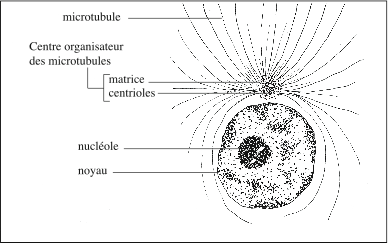 |
| Cellule en interphase. Le centre organisateur des microtubules est un lieu de nucléation à partir duquel se forment les microtubules. Ils s’accrochent au centrosome par leur extrémité (-) et s’allongent par leur extrémité (+) en recrutant les molécules de tubuline présentes dans le cytosol. Le réseau de microtubules (il n’y en a généralement qu’un seul par cellule) irradie dans tout le cytoplasme. Pour se déplacer, les organites subcellulaires se fixent sur des protéines motrices liées aux microtubules. |
Dans la cellule en interphase, le centrosome organise l’assemblage des molécules de tubuline en microtubules qui servent de « rails » aux vésicules assurant le transport antérograde de protéines et de lipides du réticulum endoplasmique à la membrane plasmique, et le transport vésiculaire rétrograde de la membrane plasmique vers les compartiments membranaires internes. Microtubules, filaments d’actine et filaments intermédiaires constituent le cytosquelette qui confère forme et polarité à la cellule et qui, dans certains cas, lui permet de se déplacer.
Nucléine, filaments, noyau
Friedrich Miescher fut le premier à montrer que la nucléine résiste aux enzymes protéolytiques. Cette observation fut confirmée : le contenu des noyaux n’est pas affecté par un traitement à la pepsine comme l’ont montré Eduard Zacharias, directeur du Jardin botanique de Hambourg, pour les noyaux d’érythrocytes de crapaud ou de protozoaires ciliés (Paramecium, Vorticella), et Eduard Strasburger, pour les Kernplatten-elemente des cellules végétales ; l’affinité pour les colorants basiques est conservée. Zacharias a montré que le contenu des noyaux disparaît après hydrolyse en milieu alcalin. Daniel Mazia (Department of Zoology, University of California, Berkeley) nota que le traitement par la pepsine fait disparaître une partie du matériel des chromosomes géants des glandes salivaires de diptères (visibles au microscope optique), sans affecter leur affinité pour leur réactif de Feulgen.
Références : Zacharias E Ueber die chemische Beschaffenheit des Zellkerns (1881)
Mazia D, Jaeger L Nuclease action, protease action and histochemical tests on salivary chromosomes of Drosophila (1939)
Il était indispensable d’établir avec certitude la localisation subcellulaire de l’ADN – seulement dans le noyau ? – si l’on voulait comprendre sa fonction et celle du noyau ; ce fut le sujet de la dissertation doctorale en chimie de Tjorbörn O. Caspersson (Nobel Institute for Cell Research, Karolinska Institutet, Stockholm). L’examen de coupes de tissus biologiques au microscope optique équipé d’un spectrophotomètre, en lumière ultraviolette, à 260 nanomètres (pic d’absorption de l’ADN et de l’ARN), et en lumière visible, après réaction de Feulgen qui ne colore que l’ADN, l’amena à conclure que l’ADN est présent dans le noyau mais que le cytoplasme contient aussi des acides nucléiques. En 1933, Jean Brachet (Université libre de Bruxelles) avait conclu que la basophilie du cytoplasme est due à la présence d’acide zymonucléique (ARN) ; la basophilie disparaît après traitement à la ribonucléase. En équipant le spectrophotomètre d’une optique en quartz (n’absorbant pas le rayonnement ultraviolet), Caspersson pouvait mesurer la quantité d’acides nucléiques. C’est ainsi qu’il mit en évidence le doublement de la quantité d’ADN avant la division cellulaire (dû au doublement du nombre de chromosomes pendant la prophase de la mitose).
Références : Caspersson TO Über den chemischen Aufbau der Strukturen des Zellkernes (1936)
Caspersson TO, Schultz J Pentose Nucleotides in the Cytoplasm of Growing Tissues (1939)
Brachet J Recherches sur la synthèse de l’acide thymonucléique pendant le développement de l’œuf d’oursin (1933)
Brachet J La localisation de l’acide thymonucléique pendant l’oogénèse et la maturation chez les Amphibiens (1940)
Brachet J La localisation des acides pentosenucléiques dans les tissus animaux et les œufs d’Amphibiens en voie de développement (1941)
Les premiers travaux sur la purification des noyaux remontent au XIXe siècle (Friedrich Miescher, 1869, Burton, 1870). La nature du matériel génétique (protéines, acides nucléiques ?) a longtemps été matière à conjectures. La purification des noyaux se heurtait deux contraintes : (i) pousser au maximum le processus de purification afin d’éviter la contamination par des protéines cytoplasmiques ; (ii) préserver l’intégrité de l’enveloppe nucléaire pour éviter la perte de nucléoplasme. En 1932, Martin Behrens mit au point une méthode en quatre étapes : (i) congélation rapide des tissus ; (ii) lyophilisation ; (iii) homogénéisation ; (iv) fractionnement en gradient de densité de solvants organiques. Le résultat fut décevant, en partie à cause de l’effet destructeur des solvants organiques sur l’enveloppe nucléaire (solubilisation des phospholipides). Behrens abandonna ses travaux en 1938 et le problème resta en suspens jusqu’aux années 1950. R. M. Schneider et M. L. Peterman montrèrent que la présence de cations divalents (calcium) dans le milieu d’homogénéisation préserve l’intégrité de l’enveloppe nucléaire et maintient son imperméabilité aux macromolécules. Plusieurs méthodes de purification des noyaux furent proposées, notamment par Vincent G. Allfrey (Rockefeller Institute for Medical Research), Alexander L. Dounce (Department of Biochemistry and Pathology, University of Rochester), Günther Siebert (Institut für Biologische Chemie und Ernährungswissenschaft, Universität Hohenheim). Divers milieux et conditions d’homogénéisation furent essayés : solution d’acide citrique, de saccharose, Waring Blender à couteaux d’acier, homogénéiseur en verre avec un piston en téflon, homogénéiseur en verre. Le contrôle de qualité des noyaux par examen au microscope électronique fut introduit, en 1960, par Georgii P. Georgiev et Yu S. Chentsov (Engelhardt Institute of Molecular Biology, USSR Academy of Sciences, Moscow). L’analyse de ces préparations purifiées permit d’établir la composition biochimique des noyaux d’une variété de tissus (foie, rein, thymus, rate cerveau, muscle) et de montrer que le noyau contient de l’ADN et de l’ARN.
Références : Behrens M Untersuchungen an isolierten Zell-und Gewebsbestandteilen. I. Mitteilung: Isolierung von Zellkernen des Kalbsherzmuskels (1932)
Behrens M Zell-und Gewebetrennung (1938)
Allfrey V, Stern H, Mirsky AE, Saetren H The isolation of cell nuclei in non-aqueous media (1952)
Dounce AL The Isolation and Composition of Cell Nuclei and Nucleoli (1955)
Dounce AL The isolation of nuclei from tumor cells (1963)
Schneider RM, Peterman AL Nuclei from normal and leukemic mouse spleen: I. The isolation of nuclei in neutral medium (1950)
Siebert G, Langendorf H Ionenhaushalt im Zellkern (1970)
Les cellules eucaryotes possèdent un ou plusieurs noyaux, à l’exception des globules rouges humains qui n’en ont pas. La destruction au laser de cet organite ou son extraction par micromanipulation entraînent la mort rapide de la cellule. Le bon fonctionnement du noyau est indispensable à la survie de la cellule ; il assure : (i) la ségrégation et le stockage du matériel génétique ; (ii) la duplication du matériel génétique avant la division cellulaire ; (iii) la copie du génome en ARN. Le noyau possède l’équipement enzymatique nécessaire à la réplication du matériel génétique et à son expression. L’expression du génome comporte deux phases : (i) une étape intranucléaire de transcription des gènes en ARN (ARNm, ARNt, ARNr et ARN de petite taille) ; (ii) une étape extranucléaire de traduction de l’ARNm et de synthèse des protéines par les ribosomes cytosoliques libres ou liés au réticulum endoplasmique. Les produits de traduction – protéines de structure et enzymes – assurent le fonctionnement, la différenciation, la division des cellules et leur adaptation aux variations de l’environnement. Les échanges entre nucléoplasme et cytoplasme se font à travers les pores de l’enveloppe nucléaire : les ARN et les ribonucléoprotéines (en premier lieu les sous-unités ribosomiales) migrent du noyau vers le cytoplasme, tandis que les ADN polymérases, ARN polymérases, protéines télomériques, histones et enzymes divers, synthétisées par les polysomes libres, migrent du cytosol vers le nucléoplasme. Chez les Ciliés, des êtres unicellulaires appartenant au groupe des Protistes, les fonctions de stockage et de duplication du matériel génétique sont assurées par le micronucleus, et la synthèse des ARN se déroule dans le macronucleus. Chez les procaryotes, dépourvus de noyau, transcription et traduction se déroulent dans le compartiment cellulaire.
Le noyau est le dépositaire du patrimoine génétique ; aujourd’hui, personne ne songerait à le mettre en doute ; ce ne fut pas toujours le cas. Noyau ou cytoplasme ? la question fit l’objet d’âpres débats. Carl von Nägeli (professeur de Botanique, École Polytechnique Fédérale et Université de Zurich), élève de Lorenz Oken – le pape de la Naturphilosophie – et de Mathias Schleiden, soutenait que « le transmetteur hypothétique des caractères héréditaires » est l’« idioplasme », une portion particulière du cytoplasme qu’il ne parvint jamais à définir avec précision. J’ajoute que Nägeli croyait aussi à la génération spontanée ! Dans les années 1760, au cours d’expériences d’hybridation avec diverses espèces de plants de tabac (Nicotiana rustica, Nicotiana paniculata), le botaniste Joseph G. Kölreuter avait conclu que l’ovule et le spermatozoïde contribuent de manière égale à la transmission des caractères héréditaires. Utilisant, comme le firent Oscar Hertwig ou Hermann Fol, l’argument des tailles respectives du cytoplasme de l’œuf et du spermatozoïde, il estimait que la transmission de l’hérédité ne pouvait pas incomber au cytoplasme ou, du moins, pas à sa totalité. Parmi un fatras de spéculations plus fantaisistes les unes que les autres, Ernst Haeckel (professeur d’Anatomie comparée, Institut de Zoologie, Université d’Iéna), propagateur du Darwinisme en Allemagne, tranchait en faveur du noyau, une opinion que partageait et soutenait Wilhelm Roux (professeur à l’Institut d’Embryologie et de Mécanique du Développement, Université de Breslau). Eduard Strasburger apporta en faveur de cette thèse un argument expérimental tiré de l’observation de la fécondation de l’orchidée : après s’être frayé un chemin à travers le pistil d’Orchis latifolia, le tube pollinique, parvenu au contact du sac embryonnaire, éjecte un noyau.
Oskar W.A. Hertwig (Medizinischen Fakultät für Anatomie, Jena, puis Anatomische Institut, Berlin) fut le premier à dire que la nucléine de Friedrich Miescher est le matériel héréditaire (1884-1885). A l’époque où Hertwig entreprend ses recherches sur le processus de la fertilisation, l’une des théories en vogue est que le spermatozoïde entre en contact avec l’œuf et lui transmet des « vibrations », ce qui déclenche le processus. Hertwig va décrire le processus réel après avoir découvert l’usage que l’on peut faire de l’œuf d’oursin comme matériel expérimental ; sa transparence lui permit d’observer au microscope l’entrée d’un seul spermatozoïde dans l’œuf et sa fusion avec le noyau. Cyrus H. Fiske et Yellagaprada Subbarow, dans le département d’Otto Folin (Harvard Medical School), et H. Karl H.A. Lohman (assistant d’Otto F. Meyerhof, puis professeur de Chimie Physiologique, Humbold Universität, Berlin) montrèrent que le phosphate présent dans le tissu musculaire appartient à des composés phosphorés : l’adénosine triphosphate, ou ATP, et la phosphocréatine. Le rôle de l’ATP – que Lohman appelait « adénylpyrophosphate » – fut décrit par lui en 1934. Le rôle de l’ATP dans les échanges énergétiques fut élucidé par Otto H. Warburg, Fritz Meyerhof et Fritz A. Lipmann, en 1937 et 1938. L’ADN contient un nucléotide apparenté à l’ATP ; on lui attribua la fonction de réserve de nucléotides à usage énergétique pour les cellules.
Un autre débat opposa les tenants de la « division directe » du noyau par étranglement, comme Rudolf Virchow ou Robert Remak, à ceux de la « division indirecte », avec partition longitudinale et migration des chromosomes. Wilhelm Roux (professeur d’Anatomie, Universität Innsbruck) choisit la seconde option en faisant valoir que le matériel nucléaire, tel qu’on peut l’observer au microscope, est réparti de manière hétérogène ; dans le cas de la division directe, cette hétérogénéité empêcherait une égale répartition quantitative et qualitative du matériel héréditaire.
Références : Kölreuter JG Vorläufige Nachricht von einigen, das Geschlecht der Pflanzen betreffenden Versuchen und Beobachtungen (1761)
Haeckel E Allgemeine Anatomie (1866)
Fiske CH, SubbaRow Y The Colorimetric Determination of Phosphorus (1925). Cet article est l’un des plus cités de la littérature scientifique.
Fiske CH, SubbaRow The Nature of the “Inorganic Phosphate” in voluntary muscle (1927)
Fiske CH, SubbaRow Y Phosphorus Compounds of Muscle and Liver (1929)
Lohmann K Über die Pyrophosphatfraktion im Muskel (1929)
Des « facteurs particulaires » aux chromosomes
« … de ces recherches,… il découle au moins un résultat certain, et ce résultat, c’est l’existence d’une substance héréditaire, d’un véhicule matériel des tendances héréditaires, et le fait que cette substance est contenue dans le noyau de la cellule germinative et dans cette partie du filament nucléaire qui, à certains moments, revêt la forme d’anses ou de baguettes courtes. »
Auguste Weismann
L’histoire de la découverte de Johan Mendel (Gregor en religion) est connue. En bref, il entreprit en 1856 la culture de pois dans le jardin botanique de son monastère, à Brno, en Autriche, et fit une étude statistique du mode de transmission d’une génération à la suivante de caractères organoleptiques « dominants » (graines lisses, graines de couleur jaune, etc…) et « récessifs » (graines ridées, graines de couleur verte, etc…). Les résultats de ces travaux l’amenèrent à formuler l’hypothèse que les caractères héréditaires sont déterminés par des « unités discrètes », des « facteurs particulaires », transmissibles individuellement des parents à leur descendance, en conservant leur intégrité. Il nota que les caractères récessifs disparaissent chez les hybrides de première génération mais réapparaissent chez les hybrides de seconde génération. Mendel présenta ses résultats en février et en mars 1865 aux membres de la Société d’histoire naturelle de Brünn et publia, en 1866, un mémoire dans les Verhandlungen des Naturforschenden Vereines in Brünn.
A propos de la découverte Mendel, deux questions se posent : (i) Pourquoi a-t-il réussi à établir les lois de transmission des caractères organoleptiques chez les plantes là où tant d’autres avant lui avaient échoué ? Pour ne citer que lui, le botaniste Josef G. Kölreuter (Académie Impériale des Sciences, Saint-Pétersbourg) avait, dans les années 1760, mené sans réel succès de nombreuses expériences d’hybridation avec Nicotiana. (ii) Pourquoi la publication de 1866 tomba-t-elle dans l’oubli pendant trente-cinq ans ? A la première question, l’on peut répondre que le succès de Mendel tient en grande partie au choix judicieux, et en partie involontaire, du matériel expérimental : des variétés pures de pois, obtenues après une série d’autofécondations. Les descendants étaient semblables aux parents et ne variaient entre eux que par un nombre limité de caractères organoleptiques présents sous deux formes : graine lisse ou graine ridé, graine jaune ou graine verte, etc, ce qui permettait de suivre aisément le passage d’un caractère d’une génération à la suivante. En outre, un heureux hasard voulut que les gènes étudiés par Mendel soient localisés sur des chromosomes différents. Enfin, en suivant à l’Université de Vienne les cours de statistiques du physicien et mathématicien Christian J. Doppler, Mendel fut en mesure de présenter ses résultats de manière analytique. Il est plus difficile d’apporter une réponse à la seconde question. Lorsqu’en 1865 Mendel présenta ses résultats à la Société d’histoire naturelle de Brünn, il est plus que probable que rares furent les membres qui en comprirent la portée. Mendel envoya des copies de son mémoire à des botanistes de renom parmi lesquels Carl W. von Nägeli (Université de Munich) ; celui-ci lui envoya une lettre de remerciement en lui promettant de procéder à une vérification expérimentale de ses conclusions. Cette belle promesse resta lettre morte. Au moment où il reçoit le mémoire de Mendel, Nägeli rédige un ouvrage (paru en 1884) exposant sa fumeuse théorie mécanico-physiologique de la descendance et sa non moins fumeuse conception du rôle de l’« idioplasme » dans la transmission des caractères héréditaires.
Les lois de Mendel s’appliquent à la répartition des caractères héréditaires chez les plantes. Plus tard, William Bateson (University of Cambridge), le promoteur de l’emploi du terme « génétique », et Lucien C.M.J. Cuénot, (professeur de Zoologie à l’Université de Nancy) montrèrent qu’elles s’appliquent aussi au monde animal. Ces lois furent redécouvertes à la fin du XIXe siècle. Hugo M. de Vries (professeur de Botanique, Université d’Amsterdam) entreprit, en 1880, des travaux sur l’hérédité des plantes (œnothère, Lychnis, Solanum, Datura, Chélidoine, Pavot, …). En 1859, Charles Darwin avait fait paraître chez l’éditeur John Murray, à Londres, son célèbre ouvrage sur l’origine des espèces. Selon lui, l’évolution était un phénomène lent, graduel, progressif : « la nature ne fait pas de sauts », avait-il coutume de dire. Ce n’est pas tout à fait exact… elle en fait parfois, comme allait le montrer de Vries avec la découverte des « mutations ». Comme Mendel, de Vries fit le choix du bon matériel expérimental : l’œnothère à grandes fleurs ou œnothère de Lamarck (Enothera lamarckiana). Parmi les milliers de plants de primevère mis en culture, il observa l’apparition d’un petit nombre d’individus (moins de 1/1.000) dont les caractères organoleptiques étaient différents de ceux de leurs parents : des plantes naines ou à grandes feuilles. Ces individus, mis en culture, transmettaient les nouveaux caractères à leur descendance. De Vries aboutit à la conclusion que, lors du passage d’une génération à la suivante, des variations brusques affectent la transmission à la descendance de « particules élémentaires d’hérédité », qu’il appela « pangènes » ; il donna à ces changements le nom de « mutations » et proposa une vision de l’évolution faisant intervenir ces brusques variations dans un ouvrage qui parut au début du XXe siècle. Ses résultats firent l’objet de deux articles publiés en 1900 ; le premier, en français, ne faisait pas référence à Mendel. Cette omission fut corrigée dans le second article, en allemand. Ce comportement vertueux fut peut être inspiré à de Vries par la lecture de l’article publié en 1900 par Carl F.J.E. Correns (Universität Tübingen), et dont le titre comportait le nom du découvreur des lois de l’hérédité. Lorsqu’il était étudiant à l’Université de Munich, Correns a eu von Nägeli comme professeur ; ce dernier lui a-t-il signalé le mémoire du moine de Brno dont il avait reçu une copie… trente-cinq ans plus tôt ? Enfin, un troisième botaniste, le jeune Erich von Tschermak-Seysenegg (Académie d’Agriculture de Vienne), cita la publication de Mendel dans sa thèse de doctorat soutenue en 1900 ; son grand-père, l’illustre botaniste Eduard Fenzl, avait eu Johan Mendel comme étudiant.
Note. En 1900, Tschermak, le benjamin, a 29 ans, Correns, 36 ans et de Vries, 52 ans.
Références : Mendel G Versuche über Pflanzenhybriden (1866)
von Nägeli CW Mechanisch-physiologische Theorie der Abstammungslehre (1884)
Darwin C On the origin of species by means of natural selection, or preservation of favoured races in the struggle for life (1859)
de Vries HM Intracellulare Pangenesis (1889)
de Vries HM Die Mutationstheorie (1901, 1903)
de Vries HM Sur la loi de disjonction des hybrides (1900)
de Vries HM Das Spallungsgezetz der Bastarde (1900)
Correns CFJE G. Mendel’s Regel über das Verhalten der Nachkommenschaft der Rassenbastarde (1900)
Certains considèrent A. Friedrich L. Weismann (Institut für Zoologie, Universität Freibourg) comme l’évolutionniste le plus important du XIXe siècle après Charles Darwin. Plus théoricien que expérimentateur (il perdit progressivement la vue), Weissman proposa, en 1885, une théorie de la transmission des caractères héréditaires qui attribue un rôle majeur aux cellules germinales ; le « plasma germinatif » (Keimplasma) du spermatozoïde et de l’œuf constitue le matériel héréditaire et il est localisé le noyau. Le travail de Walter S. Sutton, un brillant étudiant de Columbia University, portait sur les cellules somatiques. Dans les deux articles qu’il publia en 1902 et 1903 il établit un lien entre les lois de Mendel et l’hypothèse chromosomique de l’hérédité en montrant qu’il existe des similitudes entre le comportement des chromosomes au cours de la division cellulaire et celui des facteurs mendéliens de l’hérédité.
Clarence E. McClung (Professor of Biology, University of Pennsylvania) utilisait comme matériel expérimental des sauterelles ; ces insectes ont des spermatogonies de grande taille, faciles à observer au microscope optique ; les chromosomes, visibles lors de la division cellulaire, sont facilement reconnaissables individuellement. A partir de ce matériel, McClung identifia un chromosome présent dans la moitié seulement des cellules germinales ; il en déduisit que ce chromosome devait déterminer le sexe ; pour la première fois, un lien était établi entre un chromosome et un caractère phénotypique. En 1898, Sutton entreprit sa thèse de doctorat sous la direction de McClung, sur la formation des gamètes chez la sauterelle Brachystola magna. Dans l’épithélium séminifère, les spermatogonies (diploïdes) se transforment successivement en spermatocytes, spermatides et spermatozoïdes (haploïdes). Brachystola possède onze chromosomes morphologiquement différents, dont le chromosome accessoire de McClung. Ces différences de morphologie traduisaient des différences qualitatives entre les chromosomes. Sutton poursuivit son travail dans le laboratoire d’Edmund B. Wilson (Professor of Zoology, Columbia University), co-découvreur avec Nettie Stevens (Bryn Mawr College) des chromosomes sexuels XY, chez les mâles, et XX, chez les femelles.
Sutton montra que le matériel génétique des cellules somatiques de Brachystola, se présentent sous forme de paires de chromosomes homologues ; ils gardent leur intégrité à travers les divisions cellulaires et se répartissent de manière indépendante dans les cellules filles. Sutton avança l’idée que les « facteurs particulaires » de Mendel (que Sutton appelait « gènes ») sont localisés sur les chromosomes. Les travaux de Sutton sur la méïose dans les cellules germinales en font le pionnier de la cytogénétique, une branche de la biologie appelée à un grand développement au cours du XXe siècle. Il montra que la répartition des chromosomes parentaux entre les cellules germinales, leur association par paires et leur séparation au cours de la réduction chromatique corroborent les conceptions mendéliennes sur la ségrégation des caractères héréditaires ; elles seraient l’expression physique de la manière aléatoire dont se répartissent les gènes entre cellules filles. Alors qu’il semblait promis à une brillante carrière universitaire, Sutton abandonna la recherche – il n’acheva pas son doctorat -, se tourna vers les études de médecine et devint chirurgien à Kansas City. On ne peut que le regretter. Cruelle ironie du sort pour un chirurgien : il mourut à 39 ans des suites d’une banale appendicite. Sa théorie chromosomique de l’hérédité était prémonitoire et rejoignait celle formulée par Theodor Boveri à la suite de ses travaux sur l’embryogenèse de l’œuf d’oursin. Pourtant, l’accueil dans les milieux scientifiques de ce que nous appelons aujourd’hui la « théorie chromosomique de Sutton-Bovery » fut mitigé pour ne pas dire sceptique ; Wilson lui-même eut du mal à la comprendre et à en apprécier la portée. En 1902, Sutton avait reçu une leçon de mendélisme lors du passage à New York de William Bateson (John Innes Institute, London). Surnommé « The great prophet of Mendelism », Bateson avait traduit en anglais la publication de Mendel et s’était fait le propagandiste de la théorie mendélienne de l’hérédité au cours de conférences et de tournées des laboratoires. Faisant preuve d’un surprenant manque de clairvoyance, Bateson et Thomas Hunt Morgan furent parmi ceux qui rejetèrent le plus catégoriquement la théorie de Sutton.
Références : Weismann AFL Die Continuität des Keimplasmas als Grundlage einer Theorie der Vererbung (1885)
McClung CE The accessory chromosome-sex determinant (1902)
Stevens NM Studies in Spermatogenesis, with Especial Reference to the ‘Accessory Chromosome’ (1905)
Stevens NM (1906) Studies in Spermatogenesis Part II, A Comparative Study of Heterochromosomes in certain Species of Coleoptera, Hemiptera, and Lepidoptera with Especial Reference to Sex Determination (1906)
Sutton WS On the Morphology of the Chromosome Group in Brachystola magna (1902)
Sutton WS The chromosomes in heredity (1903)
Note : L’article publié en 1903 par Walter Stanborough Sutton est devenu un « classique » de la Génétique.
Les erreurs innées du métabolisme
Archibald E. Garrod (Regius professor of Medicine, University of Oxford) était un brillant clinicien : il accomplit une œuvre remarquable en établissant le lien entre facteurs particulaires mendéliens (les gènes) et enzymes. Garrod n’était pas destiné à devenir médecin comme son père ; celui-ci souhaitait le voir faire carrière dans les affaires mais devant les aptitudes intellectuelles du jeune étudiant ses maîtres l’orientèrent vers la faculté de médecine de l’Université d’Oxford. Garrod père s’était distingué dans l’étude de la goutte en utilisant une approche chimique pour comprendre le mécanisme d’accumulation d’acide urique dans les articulations. Garrod fils lui emboîta le pas en s’intéressant à une maladie héréditaire au cours de laquelle l’urine des patients, exposée à l’air, prend une coloration rouge foncé. Les sujets développent une forme d’arthrite invalidante et sont atteints d’une anomalie métabolique impliquant l’acide aminé tyrosine ; chez le sujet normal, son catabolisme conduit à la formation d’alcaptone ou acide homogentisique ; chez les patients, ce catabolite est produit à un taux anormalement élevé et passe du sang dans l’urine des patients ; son oxydation est responsable de la coloration des urines. Garrod collecta les urines des patients et des membres de leur entourage, nota soigneusement leurs caractères organoleptiques et se livra à des enquêtes familiales (on dirait aujourd’hui des enquêtes épidémiologiques). En 1902, il publia ses résultats dans The Lancet. Converti au Mendélisme par William Bateson, Garrod conclut que l’alcaptonurie est une maladie héréditaire récessive dans laquelle un enzyme du métabolisme de la tyrosine est défectueux. La maladie affecte les individus homozygotes pour l’allèle (récessif) du gène. Trop audacieuse pour l’époque, cette hypothèse ne rencontra que peu d’échos dans la communauté scientifique. Loin de se décourager, Archibald Garrod étudia d’autres maladies métaboliques héréditaires, comme la cystinurie, la pentosurie, et l’albinisme. Dans son célèbre ouvrage, publié en 1923 (une réédition de son article de 1909), il définit un champ nouveau de la pathologie : celui des erreurs innées du métabolisme. Il formula l’hypothèse que les gènes ont un impact biochimique en contrôlant l’activité des enzymes. La mutation d’un gène peut entraîner l’inactivation ou l’absence de l’enzyme codé par ce gène, ou l’altération de son fonctionnement normal.
Références : Garrod AE The Incidence of Alkaptonuria : A Study of Chemical Individuality (1902)
Garrod AE Inborn errors of metabolism (1909, 1923)
Garrod AE The Inborn Factors of Disease (1931)
Thomas H. Morgan : Naissance de la Génétique expérimentale
Au début de sa carrière universitaire, Thomas Hunt Morgan était un mutationniste dans la ligne d’Hugo de Vries. Les idées de ce botaniste, qui découvrit en 1900 la publication de Mendel, allaient dans le sens de la théorie de l’évolution en offrant une explication à l’apparition de nouvelles espèces. Par contre, Morgan affichait un certain scepticisme vis à vis de la théorie mendélienne sur la transmission des caractères héréditaires et à fortiori, comme je l’ai dit plus haut, de la théorie chromosomique de Sutton-Bovery. C’est donc dans un esprit critique qu’il décida d’examiner les conclusions d’Archibald Garrod sur la relation supposée entre gènes et enzymes.
A la base du succès de Morgan, il y a le choix d’un matériel expérimental simple, particulièrement bien adaptée à l’établissement de corrélations entre caractères phénotypiques et gènes : la mouche du vinaigre, Drosophila melanogaster, avait déjà fait son entrée dans la panoplie expérimentale sous l’égide de Charles W. Woodworth (Entomology Department, University of California, Berkeley). Drosophila a un génome de taille modeste réparti sur quatre chromosomes ; son cycle de reproduction est de dix jours, sa descendance est nombreuse et son élevage au laboratoire peu coûteux : des bananes écrasées en guise de nourriture, des bouteilles de lait vides en guise de cages. On raconte que les bouteilles de lait déposées devant le porche des maisons se trouvant sur le trajet des Morgan Raiders – c’est ainsi que furent baptisés les collaborateurs de Morgan : Alfred Sturtevant, Hermann J. Muller et Calvin Bridges – avaient une fâcheuse tendance à disparaître. Autre avantage de Drosophila comme matériel expérimental : la présence dans les glandes salivaires de chromosomes polytènes géants qui, après coloration, montrent sur toute leur longueur une succession de bandes sombres visibles au microscope. La perte d’un gène – un phénomène appelé « délétion » – se traduit par la disparition d’une bande correspondant à la position du gène sur le chromosome. Enfin, chez la drosophile mâle, la méiose se fait sans échange de matériel génétique entre chromosomes, une particularité que Morgan ignorait mais qui facilita considérablement l’interprétation des résultats.
Drosophila melanogaster a, comme son nom l’indique, un ventre (gâster) noir (melanos) et des yeux rouges. La découverte, en 1910, du mutant White aux yeux blancs fut le point de départ d’une étude systématique des mutations affectant la couleur de l’œil. En croisant un mâle aux yeux blancs avec une femelle aux yeux rouges, Morgan établit que le caractère muté « yeux blancs » est récessif et le caractère sauvage « yeux rouges », dominant. Le croisement de femelles « yeux blancs » avec des mâles « yeux rouges » donnait naissance à des descendants dont seuls les mâles avaient les yeux blancs. Cette observation suggérait une relation entre le phénotype « couleur de l’œil » et le sexe de la mouche. En supposant l’existence d’un gène « white », Morgan établit une règle selon laquelle les gènes sont nommés d’après le phénotype des allèles mutants. Dans la « pièce des mouches » du laboratoire de Columbia University où Morgan, à l’invitation d’Edmund B. Wilson, occupait la chaire de Zoologie expérimentale, les travaux avancèrent à un rythme soutenu. La transmission des caractères phénotypiques de milliers de Drosophila fut examinée en détail. Plus de 85 mutations furent localisées sur le chromosome sexuel.
Morgan a expérimentalement établi que les caractères phénotypiques sont déterminés par les gènes (les facteurs particulaires mendéliens) ; que les gènes sont localisés sur les chromosomes (ce que Walter Sutton avait déjà montré chez la sauterelle) ; que chaque chromosome contient un groupe déterminé de gènes. Morgan formula l’hypothèse que les gènes sont alignés linéairement sur les chromosomes comme les grains d’un chapelet, selon un ordre immuable. Après avoir mis au point la recombinaison chromosomique, une méthode permettant de déterminer la position d’un gène sur un chromosome, Morgan et Sturtevant établirent des cartes génétiques donnant la position de chaque gène étudié sur son chromosome. Ils calculèrent la distance séparant deux gènes contigus à partir des fréquences de recombinaison. Finalement, la position de centaines de gènes fut déterminée sur chacun des quatre chromosomes de la mouche du vinaigre. En 1912, Morgan publia les bases expérimentales de la théorie chromosomique formulée par Sutton et Boveri dans le Journal of the Academy of Natural Sciences of Philadelphia. Son ouvrage, paru en 1915, consacra l’avènement du gène comme unité fondamentale et indivisible de l’hérédité. Archibald Garrod avait établi une relation entre gène et enzyme par déduction logique ; Morgan démontra cette relation à partir de données expérimentales. Le prix Nobel de physiologie ou médecine fut attribué à Thomas Morgan en 1933, et à Hermann Muller en 1946, pour avoir découvert, en 1927, que l’exposition aux rayons X augmente considérablement le taux de mutations dans le génome de Drosophila.
Références : Morgan TH What are “factors” in Mendelian explanations? (1909)
Morgan TH Sex-limited inheritance in Drosophila (1910)
Sturtevant AH The linear arrangement of six sex-linked factors in Drosophila as shown by their mode of association (1913)
Morgan TH The mechanism of mendelian heredity (1915)
Morgan TH The Physical Basis of Heredity (1919)
Sturtevant AH The effects of unequal crossing-over at the Bar locus in Drosophila (1925)
Muller HJ Artificial transmutation of the gene (1927)
Muller HJ Types of visible variations induced by X-rays in Drosophila (1930)
Beadle et Tatum : « One gene one enzyme »
Plus de trois décennies après la publication d’Archibald Garrod sur l’alcaptonurie, le généticien George W. Beadle, un élève de Thomas Morgan, et le microbiologiste Edward L. Tatum, tous deux à Stanford University, confirmèrent expérimentalement l’hypothèse de Garrod sur la relation entre gènes et enzymes. Le champignon filamenteux microscopique Neurospora crassa est un matériel expérimental de choix pour les généticiens : (i) son cycle vital haploïde facilite considérablement l’analyse ; il n’y a pas de second allèle susceptible de masquer les effets de l’expression ou de la mutation d’un gène récessif ;(ii) les exigences nutritives de la « souche sauvage » de Neurospora – dite « souche prototrophe » – sont modestes : un milieu contenant une source de carbone : du glucose, une source d’azote : du chlorure d’ammonium, une vitamine et quelques minéraux ; (iii) on peut générer des mutants avec des besoins nutritionnels spécifiques en appliquant le protocole d’irradiation aux rayons X mis au point par Hermann Müller pour Drosophila.
Tatum fit une découverte cruciale pour la suite des travaux : la présence de biotine (vitamine H ou B8) est indispensable à la croissance de Neurospora. L’irradiation d’une culture du champignon permit d’obtenir des « mutants auxotrophes » qui, à la différence de la souche prototrophe, avaient perdu la capacité de pousser en « milieu minimum » ; certains avaient perdu la capacité de synthétiser un acide aminé (l’arginine), d’autres, la biotine. L’irradiation avait altéré ou supprimé des enzymes de la voie biosynthétique de l’arginine ou de la biotine. Pour relancer la croissance du mutant auxotrophe, il fallait ajouter au milieu de culture le métabolite manquant. Pour chaque mutant, Beadle et Tatum caractérisèrent le métabolite manquant, ce qui leur permit de remonter jusqu’à l’enzyme affecté par la mutation. L’article publié en 1941 établit la notoriété de Beadle et Tatum, de même que la formule lapidaire de leur hypothèse : « One gene one enzyme ». Il ne fallut que 17 ans aux membres du jury Nobel pour apprécier l’importance de leur contribution scientifique ; ils reçurent le prix Nobel de physiologie ou médecine en 1958.
Référence : Beadle GW, Tatum EL Genetic control of biochemical reactions in Neurospora (1941)
Protéine ou ADN ? Nature du matériel génétique
« … in contrast to the popular conception supported by newspapers and mothers of scientists, a goodly number of scientists are not only narrow-minded and dull, but also just stupid. »
James D. Watson
Les protéines sont faites de vingt acides aminés différents, l’ADN, de seulement quatre bases différentes ; cela semble peu pour rendre compte de la diversité génétique. La découverte, dans le noyau des cellules, de protamine – par Friedrich Miescher – et d’histone – par Albrecht Kossel- associées à la nucléine, amena nombre de scientifiques à croire que ces protéines basiques pouvaient constituer le matériel génétique.
Le pneumocoque (Streptococcus pneumoniae) fut dans le passé responsable de nombreuses et graves épidémies de pneumonie. Cette bactérie est entourée d’une capsule de polysaccharides mais il existe un mutant sans capsule dénué de pouvoir pathogène. Au Ministère de la santé, à Londres, le médecin Frederick Griffith essayait de développer un vaccin contre la pneumonie à pneumocoques. Au cours d’essais d’inoculation à des souris de bactéries inactivées par la chaleur, il injecta un mélange de mutant sans capsule et de bactéries avec capsule rendues non pathogènes par chauffage. Contrairement au résultat attendu, les souris développèrent une pneumonie mortelle et les résultats de l’autopsie montrèrent que les tissus renfermaient la forme encapsulée de la bactérie. Un « principe transformant » avait changé le mutant non pathogène en pneumocoque pathogène. Ce résultat ne manqua pas de susciter des objections de la part de la communauté scientifique qui avança entre autres arguments que la préparation de mutants était contaminée par des bactéries virulentes. Ces critiques ne découragèrent pas Oswald Theodor Avery (Rockefeller Institute for Medical Research) ; il croyait à l’hypothèse de la « transformation » formulée par Griffith et au passage de « caractères » d’une souche bactérienne à l’autre. Avec ses collaborateurs, il entreprit un laborieux travail de purification du principe transformant. Dès 1933, James Lionel Alloway, un résident en médecine, était parvenu à le solubiliser et à l’isoler partiellement. Avery poursuivit le travail de purification avec Colin Mac Leod et Maclyn McCarthy. Ils publièrent, en 1944, un article dans le Journal of Experimental Medicine dans lequel ils concluaient que le facteur transformant était l’ADN. Avery avait alors soixante-sept ans. Un prix Nobel, dont je tairai le nom, avait coutume de dire : « Personne n’est plus borné qu’un scientifique borné ». Malgré le nombre impressionnant de tests physiques, chimiques et enzymatiques auxquels le produit isolé fut soumis pour confirmer son identité, les incrédules n’en furent pas désarmés pour autant, prétendant que le principe transformant n’était pas de l’ADN mais les traces de protéines contaminant la préparation. Alfred Mirsky (The Rockefeller Institute) mit dans la balance tout son prestige de pionnier de la biologie moléculaire pour discréditer le travail d’Avery, allant jusqu’à intervenir auprès du Karolinska Institutet pour que le prix Nobel ne lui soit pas attribué.
André F. Boivin (professeur de Chimie biologique, Faculté de Médecine, Université de Strasbourg) et ses deux étudiants, Roger et Colette Vendrely, apportèrent des arguments en faveur de l’ADN. Ils montrèrent que les noyaux des cellules somatiques d’un individu contiennent tous la même quantité d’ADN ; que dans chaque espèce, la quantité d’ADN par noyau est constante et en relation directe avec le nombre de chromosomes ; que les cellules haploïdes, spermatozoïdes et ovules, contiennent une quantité d’ADN équivalente à la moitié de celle présente dans les cellules somatiques. Leurs articles, dont certains étaient rédigés en français, ne connurent pas la diffusion qu’ils méritaient ; le résultat de leurs expériences de transfert d’information génétique chez Escherichia Coli, publiés en 1947, ne put pas être reproduit, même par Boivin. Les milieux scientifiques finirent par se laisser convaincre que l’ADN – alors devenu à la mode – est le support de l’hérédité, par le résultat des expériences d’Alfred D. Hershey et de Martha Chase (Genetics Research Unit, Carnegie Institution at Cold Spring Harbor). Le bactériophage T2 est un phage lytique formé d’ADN et de protéines. Il pénètre dans Escherichia Coli en perçant la paroi de la bactérie ; la tête du phage, constituée de protéines, reste à l’extérieur ; seul l’ADN est injecté dans le bactérioplasme. La machinerie de synthèse protéique de Coli fabrique alors les protéines du phage au détriment de celles de la bactérie et produit des virions, dont l’accumulation fait éclater la paroi bactérienne ; les phages libérés infectent d’autres bactéries. Pour savoir si l’information nécessaire à la fabrication des protéines virales est transférée à Coli par une protéine du phage ou par son acide nucléique, Hershey et Chase utilisèrent des acides aminés – méthionine ou cystéine – marqués au soufre 35S et du phosphate marqué au 32P. Les bactéries furent infectées avec des phages dont les protéines étaient marqués au 35S et l’ADN, au 32P. Après infection, les phages furent détachés des bactéries par un traitement mécanique et le milieu d’incubation fut centrifugé de façon à séparer un culot de bactéries infectées et un surnageant contenant les phages. La radioactivité due au marquage par le 35S (protéines) se trouvait surtout dans le surnageant et celle due au marquage par le 32P (ADN), surtout dans le culot mais la séparation entre les deux marqueurs était loin d’être totale. Ces résultats furent publiés en 1952 dans The Journal of General Physiology. La communauté scientifique qui s’était montrée si tatillonne et si critique vis à vis des résultats d’Avery avala sans broncher ceux de Hershey et Chase. Pourtant, ils ne faisaient que confirmer, de manière plutôt moins convaincante, les résultats publiés dix ans plus tôt par Avery . Il est vrai que Alfred Hershey était, avec Max Delbrück et Salvador Luria, membre du « Groupe du phage » qui jouissait dans la communauté scientifique d’un prestige que beaucoup estiment aujourd’hui surévalué. Alfred Hershey, Max Delbrück et Salvador Luria reçurent le prix Nobel de physiologie ou médecine en 1969. Oswald Avery est le grand oublié du jury Nobel.
Note: Le « Groupe du phage » réunissait des chercheurs qui, au milieu du XXe siècle (1940 à 1960), étudiaient la génétique bactérienne et ses aspects moléculaires en utilisant les bactériophages comme matériel expérimental. Son chef de file était le physicien converti à la biologie, Max Delbrück, un élève de Niels H.D. Bohr, prix Nobel de physique 1922.
Références : Griffith F The significance of pneumococcal types (1928)
Avery OT, Mac Leod CM, McCarthy M vStudies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a deoxyribonucleic acid fraction isolated from Pneumococcus Type III (1944)
Boivin A, Vendrely R Rôle de l’acide désoxyribonucléique hautement polymérisé dans le déterminisme des caractères héréditaires des bactéries. Signification pour la biochimie générale de l’hérédité (1946)
v
Boivin A, Vendrely R On the possible role of the two nuclear acids in the living cell (1947)
Boivin A, Vendrely R L’acide désoxyribonucléique du noyau cellulaire, dépositaire des caractères héréditaires; arguments d’ordre analytiques (1948)
Vendrely R Vendrely C La teneur du noyau cellulaire en acide désoxyribonucléique à travers les organes, les individus et les espèces animales (1949)
Boivin A Directed mutation in colon bacilli by an inducing principle of desoxyribonucleic nature: Its meaning for the general biochemistry of heredity (1947)
Hershey AD, Chase M Independent functions of viral protein and nucleic acid in growth of bacteriophage (1952)
La règle de Chargaff
Erwin Chargaff (Department of Biochemistry, Columbia University) appartenait au petit groupe de scientifiques convaincus de la pertinence des résultats d’Oswald Avery. Si l’ADN est bien le dépositaire du patrimoine génétique, il existe une disproportion entre la diversité du monde vivant et la séquence tétranucléotidique répétitive du modèle de Phoebus Levene. Chargaff décida de soumettre ce modèle à l’épreuve en mesurant la teneur en bases d’acides désoxyribonucléiques de différentes sources. Un chimiste avec lequel il avait déjà collaboré, Aaron Bendich, lui suggéra d’adapter à la séparation des bases puriques et pyrimidiques la chromatographie de partage mise au point par Archer J.P. Martin (National Institute for Medical Research, London) et Richard L.M. Synge (Roswett Research Institute, Bucksburn, Scotland) pour séparer les acides aminés (Martin et Synge sont les co-lauréats du prix Nobel de chimie 1952). Chargaff et Ernst Vischer mit au point une méthode de quantification des bases par spectrophotométrie dans l’ultraviolet. Les résultats furent à la hauteur des efforts techniques consentis : (i) contrairement à ce que l’on peut prédire à partir du modèle de Levene les proportions molaires des bases varient selon les tissus analysés et selon les espèces mais pas entre individus de la même espèce ; (ii) pour tous les ADN testés, la teneur en adénine (A) est sensiblement égale à la teneur en thymine (T) et la teneur en guanine (G) égale à celle en cytosine (C) :
% A = % T
% G = % C
Dans l’ADN humain, Chargaff calcula qu’il y avait environ 30% de chacune des bases puriques A et T et 20% de chacune des bases pyrimidiques G et C. Les valeurs exactes sont : A = 30.9%, T = 29.4%, G = 19.9% et C = 19.8%. Ces résultats furent publiés en 1950 par Erwin Chargaff, Stephen Zamenhof et Charlotte Green dans la revue Nature. Tandis que la communauté des scientifiques continuait à s’interroger sur la nature du matériel génétique, en 1955, James D. Watson, dans le laboratoire de Salvador Luria (Indiana University, prix Nobel de médecine ou physiologie 1969), dont il était le premier étudiant, prit connaissance de l’article de Chargaff et en réalisa toute la portée.
Références : Gordon AH, Martin AJP, Synge RLM Partition chromatography in the study of protein constituents (1943)
Consden R, Gordon AH, Martin AJP Qualitative analysis of proteins: A partition chromatographic method using paper (1944)
Vischer E, Chargaff E The separation and characterization of purines in minute amounts of nucleic acid hydrolysates (1947)
Vischer E, Chargaff E The separation and quantitative estimation of purines and pyrimidines in minute amounts (1948)
Chargaff E, Vischer E, Doniger R, Green C, Misani F The composition of the desoxypentosc nucleic acids of thymus and spleen (1949)
Chargaff E, Zamenhof S, Green C Composition of human desoxypentose nucleic acid (1950)
Le matin des physiciens
« All we have to do was to construct a set of molecular models and begin to play… »
James D. Watson
En 1912, Max T.F. von Laue, ancien assistant de Max Planck, eut l’idée de bombarder un cristal avec un faisceau de rayons X ; leur rencontre avec les électrons des atomes du réseau produit des interférences et la formation d’une image de diffraction caractéristique. Il reçut le prix Nobel de physique 1914. William Henry Bragg et William Lawrence Bragg (Cambridge University) établirent que la diffraction se produit lorsque la longueur d’onde du rayonnement qui traverse le cristal est du même ordre de grandeur que la distance interatomique (Loi de Bragg de diffraction des rayons X), et adaptèrent la technique à l’analyse de cristaux de substances minérales. Ils se partagèrent le prix Nobel de physique 1915. La cristallographie par rayons X consiste à bombarder une cible avec un faisceau incident de rayons X très énergétiques produits par « bouffées » dans des accélérateurs de particules élémentaires appelés synchrocyclotrons (aujourd’hui, dans des synchrotrons dits de troisième génération). Comme il n’existe pas de lentilles capables de concentrer les rayons X, le faisceau diffracté ne donne pas d’image de l’échantillon étudié. Il faut recourir à une lentille virtuelle constituée d’un détecteur électronique relié à un ordinateur. L’image tridimensionnelle de l’échantillon est reconstituée par traitement mathématique des données. Dans les premiers appareils, le faisceau diffracté produisait des centaines, voire des milliers de taches sur un film photographique. La position et l’intensité de chaque tache étaient analysées et mesurées par le procédé mathématique des transformées de Fourrier et la carte de densité électronique ainsi obtenue était interprétée le plus exactement possible. L’étude théorique permettant cette interprétation fut réalisée par les Bragg et par Linus C. Pauling (Stanford University, prix Nobel de chimie 1954). Les métallurgistes utilisent la diffraction aux rayons X pour déterminer les caractéristiques d’alliages comme l’acier, et les minéralogistes, pour établir l’architecture de substances cristallines.
Références : Bragg WH An introduction to crystal analysis (1928)
Pauling L The principles determining the structure of complex crystals (1929)
La diffraction par les rayons X fut appliquée à l’étude des macromolécules biologiques dans les années 1920, dans le laboratoire de John Desmond Bernal (Department of crystallography, Cavendish Laboratory, University of Cambridge), et à l’étude des protéines, dans les années 1930, dans celui de William T. Astbury (Leeds University). Les rayons X utilisés ont une longueur d’onde proche de la distance des liaisons interatomiques dans les macromolécules biologiques : de 0,05 à 0,15 nanomètre. Le préalable à toute étude cristallographique est l’obtention d’une structure régulière et ordonnée : des cristaux (idéalement) ou des fibres orientées dans le même sens. Les premières tentatives pour obtenir des cristaux de protéines datent des années 1920 ; des cristaux d’hémoglobine et d’uréase de bonne qualité furent disponibles dans les années 1930. C’est à cette époque que William T. Astbury (Leeds University) étudia la structure, non pas de cristaux mais de fibres de collagène et de kératines, des constituants des cheveux, de la laine et de la soie. Pour interpréter ses résultats, Astbury s’inspira des travaux de Linus Pauling et Robert B. Corey sur la structure tridimensionnelle de la liaison peptidique. A leur tour, ceux-ci s’inspirèrent de la structure de la kératine publiée par Astbury pour déterminer celle de l’hélice α, la première structure protéique secondaire à être connue. Dans le laboratoire de John Bernal, au Cavendish Laboratory, Max F. Perutz un élève William Lawrence Bragg, entreprit, en 1937, l’analyse de cristaux d’hémoglobine de cheval, la protéine transportant l’oxygène dans le sang des mammifères. De 1958 à 1960, il détermina la structure tridimensionnelle de l’hémoglobine, et John C. Kendrew, celle de la myoglobine, la protéine transportant l’oxygène dans les muscles. Leurs résultats montrèrent que chaque protéine possède une structure tridimensionnelle et une conformation uniques. Cette conclusion fut confirmée par les données de David Chilton Phillips et ses collègues (Davy Faraday Research Laboratories, Royal Institution, Londres) sur le lysozyme, une protéine du blanc d’œuf. Max Perutz et John Kendrew reçurent conjointement le prix Nobel de chimie 1962.
Références : Bernal JD On the Interpretation of X-Ray, Single Crystal, Rotation Photographs (1926)
Bragg WL First stages in the X‐ray analysis of proteins (1965)
Astbury WT, Woods HJ The X-Ray Interpretation of the Structure and Elastic Properties of Hair Keratin (1930)
Astbury WT, Marwick TC X-Ray Interpretation of the Molecular Structure of Feather Keratin (1932)
Astbury WT, Woods HJ X-Ray Studies of the Structure of Hair, Wool, and Related Fibres. II. The Molecular Structure and Elastic Properties of Hair Keratin (1934)
Astbury WT, Bell FO Nature of the Intramolecular Fold in Alpha-Keratin and Alpha-Myosin (1941)
Pauling L, Corey R, Branson HR The structure of proteins; wo hydrogen-bonded helical configurations of the polypeptide chain (1951)
Pauling L, Corey R Compound Helical Configurations of Polypeptide Chains: Structure of Proteins of the α-Keratin Type (1953)
Perutz MF X-Ray Analysis of Hemoglobin (1963)
Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC A three‐dimensional model of the myoglobin molecule obtained by X‐ray analysis (1958)
Kendrew JC, Dickerson RE, Strandberg BE, Hart RG, Davies DR, Phillips DC, Shore VC Structure of myoglobin: a three‐dimensional Fourier synthesis at 2 Å resolution (1960)
Philips DC The hen egg-white lysozyme molecule (1967)
En 1937, William Astbury et la cristallographe Florence Ogilvy Bell abordèrent l’analyse cristallographique de longues fibres d’ADN fournies par Rudolf Signer (Universität Bern) et Torbjörn Caspersson (Karolinska Institutet, Stockholm). Les clichés de diffraction obtenus révélaient que l’ADN, comme les protéines, possède une structure régulière. Astbury confirma une prédiction faite par Einar Hammarsten et Signer (Department of Chemistry, Karolinska Institutet) : le plan des bases est perpendiculaire au grand axe de la molécule d’ADN et calcula la distance séparant deux bases voisines : 0,34 nanomètre, une valeur très proche de la valeur réelle dans la forme B de l’ADN. A partir de ces données rudimentaires, Astbury proposa un modèle d’ADN monocaténaire. dont s’inspirèrent Linus Pauling et Robert Corey (California Institute of Technology, Pasadena) pour proposer, en 1953, leur modèle d’ADN en triple hélice, publié dans les Proceedings of the National Academy of Sciences.
Au début des années 1950, trois groupes de chercheurs sont impliqués dans l’étude de la structure de l’ADN : le groupe de King’s College (London), Maurice H.F. Wilkins et Rosalind E. Franklin, le groupe du Cavendish Laboratory, (University of Cambridge), James D. Watson et Francis H. Crick, et le groupe de Linus Pauling au Californian Institute of Technology (Pasadena). C’est surtout entre les deux derniers groupes que la compétition est la plus serrée, le généticien Watson ayant pleinement conscience de l’impact en biologie d’une telle découverte ; en témoignent les ouvrages de Watson et de Crick parus, respectivement, en 1968 et 1988.
Rosalind Franklin avait une bonne pratique de l’analyse aux rayons X acquise durant son séjour de trois ans à Paris, au Laboratoire central des services chimiques de l’État. Rudolf Signer lui donna un sel de sodium de l’ADN, à partir duquel Wilkins obtint un précipité de fibres uniformément orientées. Franklin réalisa les clichés de diffraction à partir desquels fut introduite la distinction entre les formes dite A et B de l’ADN ; ces clichés révélaient la nature hélicoïdale de l’ADN. Watson et Crick entreprirent la construction de modèles à partir d’une série de résultats expérimentaux : (i) ceux du groupe de Bernal (Birkbeck College) qui avait établi la configuration spatiale des nucléotides, avec le plan du pentose perpendiculaire à celui de la base ; (ii) ceux contenus dans le rapport confidentiel remis par Rosalind Franklin au Medical Research Council, et qui confirmait les caractéristiques de l’hélice : diamètre : 2 nanomètres, distance entre deux bases consécutives : 0,34 nanomètre, 10 bases par tour d’hélice ; (iii) les informations fournies par le physico-chimiste Jerry Donohue sur les formes tautomériques des bases (les paires de bases sont sous la forme céto) ; (iv) la Règle de Chargaff sur l’équivalence des proportions des bases A/T (adénine-thymine) et G/C (guanine-cytosine). Enfin, dans son ouvrage paru en 1968, Watson évoque les circonstances dans lesquelles Crick et lui purent examiner, par l’intermédiaire de Maurice Wilkins, le cliché de diffraction n° 51 de la forme B de l’ADN obtenu par Franklin et son Phd student, Raymond Gosling. Watson et Crick aboutirent à la solution du problème en construisant des modèles, sans avoir fait une seule expérience de diffraction aux rayons X.
La reconstitution de la structure tridimensionnelle d’une molécule par construction de modèles physiques a été imaginée en 1928 par le chimiste Kurt Meyer (Herman Mark I.G. Farben, Ludwigshafen) pour déterminer la structure tridimensionnelle de la cellulose et de la fibroïne de la soie. Ces deux macromolécules fibrillaires présentent suffisamment d’éléments de régularité pour donner des diagrammes de diffraction interprétables. A partir du modèle physique qu’il avait élaboré, Meyer calcula les coordonnées théoriques de diffraction et les compara aux valeurs obtenues à partir de clichés cristallographiques. En 1953, Linus Pauling et Robert Corey (California Institute of Technology, Pasadena) publièrent dans les Proceedings of the National Academy of Sciences un modèle d’ADN en triple hélice, avec les groupes phosphate (chargés) dirigés vers l’intérieur et les bases (hydrophobes), vers l’extérieur de l’hélice. La « folle poursuite », pour paraphraser le titre de l’ouvrage de Francis Crick, s’acheva avec la publication, en 1953, du modèle de Watson et Crick, formé de deux chaînes poly-désoxyribonucléotidiques enroulées en double hélice droite ; les groupes phosphate sont dirigés vers l’extérieur et les bases vers l’intérieur de l’hélice. Les bases d’une des deux chaînes sont liées par des liaisons hydrogène aux bases complémentaires de l’autre chaîne : A avec T (deux liaisons) et G avec C (trois liaisons). La stabilité de la molécule est assurée par l’environnement hydrophobe créé au centre de l’hélice par les bases et par les groupes phosphates dont les charges négatives sont dirigées vers l’extérieur.
Après avoir lu beaucoup de ce qui a été écrit à propos de la découverte de la double hélice, je partage la conviction de ceux qui croient que c’est Rosalind Franklin qui a « fait l’essentiel du job ». Elle est décédée en 1958, à trente-huit ans. En 1962, presque dix ans après la publication des articles dans Nature, le jury Nobel ayant enfin compris l’importance de la découverte de la structure tridimensionnelle de l’ADN, décerna le prix de médecine ou physiologie à James Watson, Francis Crick et Maurice Wilkins.
Références : Astbury WT, Bell FO X-Ray Study of Thymonucleic Acid (1938)
Signer R, Caspersson TO, Hammarsten E Molecular Shape and Size of Thymonucleic Acid (1938)
Pauling L, Corey R A Proposed Structure for the Nucleic Acids (1953)
Watson JD The Double helix. A Personal Account of the Discovery of the Structure of DNA (1968)
Crick FH What Mad Pursuit. A personal View of Scientific Discovery (1988)
Watson JD, Crick FH Molecular Structure of Nucleic Acids; a Structure for Deoxyribose Nucleic Acid (1953)
Watson JD, Crick FH Genetical implications of the structure of deoxyribonucleic acid (1953)
Franklin RE, Gosling RG Molecular Configuration in Sodium Thymonucleate (1953)
Wilkins MHF, Stokes AR, Wilson HR Molecular Structure of Deoxypentose Nucleic Acids (1953)
Réplication
« It has not escaped our notice that the specific pairing we have postulated immediately suggests copying mechanism for the genetic material. »
James D. Watson & Francis H. Crick
Parmi les fonctions du noyau, la réplication du matériel génétique est l’une des plus importantes. Au début de la division cellulaire, les chromosomes se dédoublent et chaque cellule-fille reçoit le même nombre de chromosomes que la cellule-mère (sauf dans le cas de la méiose). Le dédoublement des chromosomes s’accompagne d’une synthèse d’ADN pendant la phase S (Synthèse) du cycle cellulaire. Dans le courant des années 1950, plusieurs mécanismes de réplication de la molécule d’ADN étaient envisageables ; dans le modèle de la double hélice, de James Watson et Francis Crick, une séquence de bases précise est nécessaire pour maintenir et perpétuer la structure hélicoïdale. Partant de cette constatation, ces auteurs suggérèrent un mécanisme de réplication dit semi-conservatif : les deux brins d’ADN se séparent et servent de matrice pour le recrutement et l’association de nucléotides complémentaires, conduisant à l’assemblage des deux brins d’une nouvelle chaîne polynucléotidique ; le choix des nucléotides est dicté par la séquence des bases du brin servant de matrice. Si ce modèle est correct, toute molécule d’ADN néo-synthétisé doit être constituée d’un brin d’ADN-matrice et d’un brin néoformé. Différentes approches expérimentales furent mises en œuvre pour le démontrer.
La première confirmation fut apportée par J. Herbert Taylor et Shirley Taylor (Department of Biological Science, The Florida State University). Ils cultivèrent des graines de haricots (Vicia faba) dans un milieu contenant les quatre nucléotides entrant dans la composition de l’ADN ; seule la thymidine était marquée au tritium (3H) ; Herbert transféra les cellules dans un milieu de culture contenant des nucléotides non marqués et de la colchicine ; cet alcaloïde, extrait de la pervenche (Vinca minor), se fixe spécifiquement sur les protéines des microtubules du fuseau mitotique, bloquant la division cellulaire au stade de la métaphase et empêchant la migration ultérieure des chromatides vers les pôles du fuseau mitotique. L’usage de l’autoradiographie à haute résolution permit d’observer la réplication semi-conservative des chromosomes condensés en chromatides. Une nouvelle confirmation fut apportée par Matthew S. Meselson et Franklin W. Stahl (California Institute of Technology). Ils firent croître Escherichia coli dans un milieu nutritif avec, comme source d’azote, du chlorure d’ammonium marqué avec l’isotope « lourd » de l’azote (15N). Après transfert des bactéries dans un milieu contenant l’isotope stable naturel (14N), un échantillon fut prélevé à intervalles réguliers ; la masse de l’ADN extrait de chaque échantillon fut déterminée par centrifugation en gradient de chlorure de césium jusqu’à l’équilibre isopycnique. La position des bandes d’ADN dans le gradient fut déterminée par lecture optique à la longueur d’onde d’absorption des acides nucléiques dans l’ultraviolet. Sur les clichés photographiques, en fonction du temps d’incubation des bactéries dans le milieu contenant 15N, apparaissait d’abord une bande « lourde » d’ADN 15N, puis une bande plus légère d’ADN hybride 15N-14N, puis une bande légère d’ADN 14N. Enfin, en 1963, John F. Cairns (John Curtin School of Medical Research, Australia et Cold Spring Laboratory) observa, par autoradiographie, la réplication semi-conservative de l’ADN circulaire d’Escherichia Coli B3 et K12 incubés pendant des durées variables en présence de [3H]thymidine.
La réplication de l’ADN est catalysée par des complexes multienzymatiques présents principalement dans le noyau : les ADN polymérases ADN-dépendantes. Celles qui ont été caractérisées sont groupées en familles (sept au total) : A, B, C, D, X, Y, RT. Les cellules procaryotes possèdent cinq polymérases, dont trois principales numérotées I, II et III, dans l’ordre de leur découverte ; les cellules eucaryotes en ont onze, dénommées α (réplication dans le noyau), β (réparation de l’ADN), γ (réplication dans les mitochondries), δ, ε, z, h, i, q, k, Rev1.
La polymérase I fut caractérisée et purifiée par Arthur Kornberg. Après ses études de médecine à Rochester University, il étudia l’enzymologie dans le laboratoire de Severo Ochoa (New York University), puis dans celui de Carl et Gery Cori, la Mecque de l’enzymologie (Washington University School of Medicine, St-Louis). En 1954, Kornberg entreprit la recherche d’enzymes catalysant la synthèse de poly désoxyribonucléotides dans des broyats d’Escherichia Coli incubés en présence de désoxyribonucléosides triphosphate marqués au 32P et de fragments d’ADN ; l’ADN polymérase qu’il identifia en 1958 sera, plus tard, baptisée ADN polymérase I. Pour vérifier qu’il s’agissait bien de la polymérase responsable de la réplication du génome d’Escherichia Coli, John F. Cairns et Paula De Lucia (Cold Spring Harbor Laboratory) mirent en contact des cultures de Coli avec un mutagène. Au prix d’un intense travail de criblage de milliers de cellules, il isolèrent un mutant répliquant son génome en l’absence de l’enzyme de Kornberg. Le gène muté fut ironiquement baptisé polA. Adoptant la stratégie de Cairn et De Lucia, Thomas B. Kornberg (le fils d’Arthur) et Malcolm L. Gefter (Department of Biological Sciences, Columbia University, New York) mirent en évidence les ADN polymérases III, puis II. Comme l’ADN polymérase I de Kornberg, l’ADN polymérase II corrige les erreurs de réplication et répare l’ADN endommagé par des agents chimiques ou physiques. L’ADN polymérase III, catalyse la réplication de l’ADN chez les procaryotes. En présence d’une matrice d’ADN, elle ajoute des nucléotides à l’extrémité 3’OH d’un ADN, ou d’une amorce d’ARN (primer), appartenant à un duplex. Reiji Okasaki et Tsuneko Okasaki (Nagoya University) montrèrent que l’ADN polymérase III ne synthétise pas les deux brins d’ADN de la même manière : le brin dit « avancé » est édifié de manière continue, nucléotide par nucléotide ; le brin « retardé » est synthétisé par morceaux qui sont ensuite assemblés et liés entre eux par l’ADN polymérase I. L’ADN polymérase III commence la réplication à partir d’un « primer » d’ARN synthétisé par une ARN polymérase. En 1994, la structure de l’ADN polymérase β fut établie par cristallographie aux rayons X, dans le laboratoire de Samuel Wilson (Laboratory of Biochemistry, National Cancer Institute), un élève de Mahlon B. Hoagland, le découvreur de l’ARNt, puis de Marshall W. Nirenberg (prix Nobel de physiologie ou médecine 1968) qui joua un rôle actif dans le déchiffrage du code génétique. Toutes les ADN polymérases ADN-dépendantes ont, depuis, été isolées, cristallisées et leur structure tridimensionnelle a été établie.
Les résultats des travaux d’Arthur Kornberg et de ses collaborateurs furent publiés en 1958. Le jury Nobel, qui commençait à comprendre l’importance de la découverte de la structure en double hélice de l’ADN, attribua dans la précipitation le prix Nobel 1959 à Kornberg et Ochoa, l’un pour la découverte de l’ADN polymérase, l’autre pour celle de l’ARN polymérase, alors que Watson et Crick attendaient toujours la reconnaissance de leur contribution. Pour la petite histoire, il faut savoir que deux articles sur l’ADN polymérase, sur les trois soumis par Kornberg au Journal of Biological Chemistry, furent rejetés par le comité de lecture dont faisait partie… Erwin Chargaff ! Il est également intéressant de savoir que l’article de Marianne Grunberg-Manago et Severo Ochoa sur l’ARN polymérase, qui valut à ce dernier l’attribution du prix Nobel, traite d’un enzyme qui synthétise des poly ribonucléotides en l’absence d’une matrice d’ADN ; lorsque cet enzyme travaille dans le bon sens, c’est une ribonucléase ! L’ARN polymérase, la vraie, fut découverte en 1959 par Samuel B. Weiss et Leonard Gladstone (Argonne Cancer Research Hospital, Department of Biochemistry, University of Chicago) dans des noyaux de foie de rat. Après les errements de 1959, le prix Nobel de chimie fut attribué, à bon escient cette fois, à Roger D. Kornberg (Stanford University), un autre fils d’Arthur, pour avoir élucidé le fonctionnement des gènes responsables de la transcription du génome en protéines dans les cellules eucaryotes, et le rôle joué par l’ARN polymérase II.
Références : Taylor JH, Woods PS, Hughes WL The organization and duplication of chromosomes as revealed by autoradiographic studies using tritium-labeled thymidine (1957)
Meselson M, Stahl FW The Replication of DNA in Escherichia coli (1958)
Lehman IR, Bessman MJ, Simms ES, Kornberg A Enzymatic Synthesis of Deoxyribonucleic Acid. I. Preparation of Substrates and Partial Purification of an Enzyme from Escherichia Coli (1958)
Bessman MJ, Lehman IR, Simms ES, Kornberg A Enzymatic Synthesis of Deoxyribonucleic Acid. II. General Properties of the Reaction (1958)
Lehman IR, Zimmerman SB, Adler J, Bessman MJ, Simms ES, Kornberg A Enzymatic Synthesis of Deoxyribonucleic Acid. V. Chemical Composition of Enzymatically Synthesized Deoxyribonucleic Acid (1958)
Grunberg-Manago M, Ortiz P, Ochoa S Enzymic synthesis of polynucleotides. I. Polynucleotide phosphorylase of Azotobacter vinelandii (1956)
De Lucia P, Cairns J Isolation of an E. coli Strain with a Mutation affecting DNA Polymerase (1969)
Kornberg T, Gefter ML Purification and DNA Synthesis in Cell-Free Extracts: Properties of DNA Polymerase II (1971)
Gefter ML, Hirota Y, Kornberg T, Wechsler JA, Barnoux C Analysis of DNA Polymerases II and III in Mutants of Escherichia Coli Thermosensitive for DNA Synthesis (1971)
Okazaki R, Okazaki T, Sakabe K, Sugimoto K, Sugino A Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains (1968)
Sugimoto K, Okazaki T, Okazaki R Mechanism of DNA chain growth. II. Accumulation of newly synthesized short chains in E. Coli infected with ligase-defective T4 phages (1968)
Weiss S, Gladstone L A Mammalian System for The Incorporation of Cytidine Triphosphate into Ribonucleic Acid (1959)
Cramer P, Bushnell D, Kornberg R Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution (2001)
Gnatt A, Cramer P, Fu J, Bushnell, Kornberg RD Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution (2001)
Colinéarité
Les années 1950 furent une période féconde pour les chimistes des protéines. On sait que les protéines sont des enchaînements linéaires d’acides aminés liés entre eux par des liaisons covalentes, selon une séquence unique et caractéristique. Frederick Sanger (Department of Biochemistry, Cambridge University et Laboratory of Molecular Biology, Medical Research Council, prix Nobel de chimie 1958) déchiffre la séquence des 51 résidus d’acides aminés des deux chaînes de l’insuline de bœuf. Stanford Moore et William H. Stein (Rockefeller University) conçoivent et développent le premier séquenceur automatique d’acides aminés, et établissent la séquence de la ribonucléase. Le groupe de Christian B. Anfinsen (Laboratory of Cellular Physiology and Metabolism, National Heart Institute, National Institute of Health, Bethesda) et celui de Moore et Stein complètent la séquence des 124 résidus d’acides aminés de la ribonucléase A de pancréas de bœuf. Des résultats de ces travaux émerge le concept de « colinéarité » que l’on peut énoncer comme suit : il existe une correspondance directe et linéaire entre la séquence des bases nucléotidiques d’un gène et celle des acides aminés de la protéine codée par ce gène. Ce concept est un jalon essentiel pour comprendre le rapport existant entre gène et enzyme, et le mécanisme de la transcription (la copie de l’ADN génomique en ARNm). Le prix Nobel de chimie 1972 fut attribué à Christian B. Anfinsen, Stanford Moore et William H. Stein.
Références : Sanger F, Tuppy H The amino-acid sequence in the phenylalanyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates (1951)
Sanger F, Thompson EO The amino-acid sequence in the glycyl chain of insulin (1952)
Hirs CH Stein WH, Moore S The amino acid composition of ribonuclease (1954)
Hirs CH Studies on the structure of ribonuclease. Enzymatic hydrolysis of the four large peptides formed in the tryptic hydrolysis of the oxidized protein (1960)
Hirs CHW, Moore S, Stein WH The Sequence of the Amino Acid Residues in Performic Acid-oxidized Ribonuclease (1960)
Smyth DG, Stein WH, Moore S The sequence of amino acid residues in bovine pancreatic ribonuclease: revisions and confirmations (1963)
Anfinsen CB, Haber E, Sela M, White Jr FH The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain (1961)
Potts JT, Berger A, Cooke J, Anfinsen CB A reinvestigation of the sequence of residues 11 to 18 in bovine pancreatic ribonuclease (1962)
Deux groupes de chercheurs vont faire la démonstration qu’il existe une colinéarité entre les séquences du gène et de la protéine. La tryptophane synthase est un enzyme de la voie biosynthétique du tryptophane, un acide aminé essentiel ; elle est composée de deux polypeptides, A et B. Au début des années 1960, Charles Yanofsky (Biological Sciences Department, Stanford University) sélectionne des formes mutées du gène de la tryptophane synthase dont il compare la séquence des bases nucléotidiques à celle des acides aminés de la protéine. Pour localiser la position des acides aminés mutés, Yanofsky utilise la technique du fingerprinting mise au point en 1956 par Vernon M. Ingram (Cavendish Laboratory, University of Cambridge) pour détecter des changements de séquence dans l’hémoglobine humaine. La chaîne A de la tryptophane synthase (268 résidus d’acides aminés) fut soumise à une digestion protéolytique ; les peptides libérés furent séparés par électrophorèse ou par chromatographie sur papier. La substitution d’un acide aminé par un autre se traduit par un changement de position sur le chromatogramme du peptide renfermant cet acide aminé. Pour déterminer la position des bases mutées, Yanofski et coll. établirent une carte transcriptionnelle du gène en adoptant la technique de recombinaison génétique mise au point, en 1946, par Joshua Lederberg et Edward L. Tatum (Yale University), tous deux prix Nobel de physiologie ou médecine 1958. J’en rappelle brièvement le principe : des bactéries Escherichia coli « mâles » font passer de l’information génétique en transférant physiquement leur chromosome (ou un chromosome auxiliaire appelé plasmide) dans des bactéries « femelles » de souche différente. Ce transfert d’information génétique peut être interrompu en divers points du gène. Il suffit de soumettre la suspension de bactéries à un traitement mécanique qui a pour effet de rompre la portion de chromosome située entre la bactérie mâle et la bactérie femelle. Mise dans des conditions de culture ad hoc, les gènes portés par les fragments de chromosomes transférés dans la bactérie réceptrice sont exprimés. En espaçant judicieusement les traitements mécaniques, on peut jalonner le transfert d’un chromosome morceau par morceau et même gène par gène. Yanofsky démontra qu’il existe une correspondance entre les mutations dans la séquence du gène de la sous-unité A de la tryptophane synthase et les substitutions d’acides aminés dans la séquence de la protéine.
Références : Ingram VM A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin (1956)
Ingram VM Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin (1957)
Lederberg J, Tatum EL Gene recombination in Escherichia coli (1946)
Lederberg J, Tatum EL Gene recombination in the bacterium Escherichia coli (1947)
Crawford IP, Yanofsky C On the separation of the tryptophan synthase of Escherichia coli into two protein components (1958)
Carlton BC, Yanofsky C Studies on the Position of Six Amino Acid Substitutions in the Tryptophan Synthetase A Protein (1963)
Carlton BC, Yanofsky C Amino Acid Substitutions in the A Proteins of Tryptophan Synthetase Mutants and Revertants (1965)
Yanofsky C, Drapeau GR, Guest JR, Carlton BC The complete aminoacid sequence of the tryptophan synthase a protein (a subunit) and its colinear relationship with the genetic map of the a gene (1967)
Guest JR, Drapeau GR, Carlton BC, Yanofsky C The Amino Acid Sequence of the A Protein (α Subunit) of the Tryptophan Synthetase of Escherichia coli. V. Order of Tryptic Peptides and the Complete Amino Acid Sequence (1967)
Yanofsky a démontré l’existence de la colinéarité gène/protéine chez les bactéries ; Vernon M. Ingram, John A. Hunt et Anthony O.W. Stretton (Cavendish Laboratory, University of Cambridge) en firent la démonstration chez les mammifères. Ingram, surnommé « The father of Molecular Medicine », entreprit, en 1955, l’étude de la drépanocytose ou anémie à cellules (hématies) falciformes, une maladie assez répandue parmi les populations des Antilles, d’Afrique, d’Inde, du Moyen-Orient et du bassin méditerranéen. Ingram montra que cette pathologie est causée par la substitution d’un seul acide aminé dans la séquence de la chaîne β de l’hémoglobine : un glutamate est remplacé par une valine. Ingram, soutenu et conseillé par Max Perutz et Francis Crick, mit cette anomalie en relation avec une mutation dans la séquence du gène.
Références : Ingram VM A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin (1956)
Ingram VM Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin (1957)
Intermédiaires : ARN messager et ARN de transfert
A la suite de la publication de l’article de Watson et Crick sur la structure en double hélice de l’ADN, Gueorgui A. Gamov (George Washington University) leur suggéra un mécanisme d’expression des gènes. Gamov était un physicien de grand renom, inventeur, avec le chanoine Georges Lemaître, du modèle cosmologique du Big Bang sur l’origine et l’évolution de l’univers. Gamov voyait la double hélice comme une matrice ou plutôt un moule comportant vingt cavités de structures différentes, dans lesquelles viennent se loger les vingt acides aminés ; cette hypothèse implique une interaction directe des acides aminés avec les bases nucléotidiques ce qui fut immédiatement écarté par Francis Crick. Mais l’hypothèse a aussi un côté prémonitoire en proposant que la sélection des acides aminés soit imposée par un « code » formé de triplets de bases nucléotidiques (64 combinaisons possibles pour 20 acides aminés). Cette idée s’imposa à Crick ; restait à en établir la réalité. Crick et Sydney Brenner s’y employèrent. La proflavine, un dérivé de l’acriflavine, exerce une action bactériostatique et mutagène en s’insérant entre les bases nucléotidiques de l’ADN, ce qui provoque des mutations par insertion ou délétion d’une paire de bases. Crick et Brenner mirent à profit ce moyen de retirer ou d’insérer des bases dans le gène rIIB du bactériophage T4 (la protéine rIIB est impliquée dans le phénomène de recombinaison génétique). Ils testèrent, sur des mutants de cultures de T4 traitées à la proflavine, l’effet des additions ou soustraction de bases sur l’expression ou l’absence d’expression du gène rIIB. Les résultats de ce travail confirmèrent que le codon à la base du code génétique est un triplet de bases.
Références : Brenner S, Benzer S, Barnett L Distribution of Proflavin-Induced Mutations in the Genetic Fine Structure (1958)
Crick FH, Barnett L, Brenner S, Watts-Tobin RJ General nature of the genetic code for proteins (1961)
Crick FHC, Griffith JS, Orgel LE Codes without Commas (1957)
La proposition de Gamov d’une lecture directe du message génétique par les acides aminés, rejetée par Crick, se heurtait au fait que, dans les cellules eucaryotes, les chromosomes sont localisés dans le noyau et les ribosomes dans le cytoplasme ; Crick et Brenner supposèrent qu’il existait au moins un intermédiaire entre le gène et la machinerie de synthèse protéique et que cet intermédiaire devait être un ARN « adaptateur » portant l’acide aminé, chacun des 20 acides aminés ayant un adaptateur spécifique. Ce postulat devint réalité à la suite des travaux de Paul C. Zamecnik, Elizabeth Keller et Mahlon Hoagland (Harvard Medical School). En 1953, ils mirent au point le premier système de synthèse protéique in vitro en incubant des homogénats de foie de rat avec des acides aminés marqués au 14C. Après injection des acides aminés 14C à des rats et fractionnement par centrifugation d’homogénats de foie, la radioactivité se retrouve initialement dans la fraction microsomes (riche en ribosomes liés aux membranes). En 1956, ils montrent que l’addition d’ATP au milieu augmente l’incorporation d’acides aminés 14C dans les protéines : l’acide aminé est « activé » en se liant à une molécule d’AMP, avec formation d’un aminoacyl adénylate. Dans le système acellulaire de synthèse protéique, en présence d’acides aminés 14C, le marquage se retrouve associé à l’ARN d’une fraction particulaire avant d’être transféré aux protéines des microsomes. Ils isolent un « ARN soluble » de petite taille, plus tard baptisé « ARN de transfert », identifiant ainsi le transporteur intermédiaire d’acides aminés dans la synthèse protéique, imaginé par Crick et Brenner. L’ARNt fixe spécifiquement, sur le ribose de son extrémité 3’, un acide aminé activé (par exemple l’alanine) en donnant un aminoacyl-ARNt (en l’occurrence l’alanyl-ARNt). Une fois chargé, l’ARNt agit comme « donneur d’acide aminé » dans la réaction d’élongation de la chaîne peptidique sur le ribosome. Il existe 20 ARNt différents spécifiques de chaque acide aminé. En 1964, Robert W. Holley détermina la séquence nucléotidique de l’alanyl-ARNt de levure. Pour la première fois, la séquence des 76 nucléotides d’un acide nucléique était connue.
Dans le système acellulaire de synthèse protéique, Hoagland et Zamecnik avaient isolé un facteur sensible à la chaleur ; il fut plus tard identifié à l’aminoacyl-ARNt synthétase, l’enzyme qui catalyse la fixation d’un acide aminé sur son ARNt. Il existe vingt aminoacyl-ARNt synthétases différentes, chacune spécifique d’un acide aminé. Ces enzymes, d’une extrême sophistication, possèdent deux sites actifs séparés, l’un reconnaissant l’acide aminé et l’autre l’ARNt correspondant. En 1941, Fritz A. Lipmann et Hermann Kalckar avaient prédit que des intermédiaires phosphorylés devaient intervenir dans la synthèse protéique. Comme je l’ai brièvement mentionné plus haut, conformément à cette prédiction, la réaction catalysée par l’aminoacyl-ARNt synthétase nécessite l’apport d’une molécule d’ATP pour « activer » le résidu d’acide aminé en le transformant en aminoacyl adénylé, c’est-à-dire lié à une molécule d’AMP.
Références : Zamecnik PC, Keller EB Relation between phosphate energy donors and incorporation of labeled amino acids into proteins (1954)
Zamecnik PC, Keller EB Relation between phosphate energy donors and incorporation of labeled amino acids into proteins (1954)
Hoagland MB, Keller EB, Zamecnik PC Enzymatic carboxyl activation of amino acids (1956)
Hoagland MB, Stephenson ML, Scott JF, Hecht LI, Zamecnik PC A Soluble Ribonucleic Acid Intermediate in Protein Synthesis (1958)
Les bactéries ont des ennemis : des virus appelés « bactériophages ». Ceux-ci ont un comportement apparemment imprévisible : après avoir infecté la bactérie en lui injectant leur ADN, (i) ils s’approprient la machinerie de transcription et de traduction de leur hôte, se multiplient et font éclater la cellule (cycle lytique) ; Seymour S. Cohen (Rockefeller Institute), un élève d’Erwin Chargaff, a montré que l’infection d’une bactérie par un phage provoque une dégradation de l’ADN bactérien et l’arrêt de la synthèse des protéines bactériennes au profit de celles du phage ; (ii) ils insèrent discrètement leur génome dans le génome bactérien et restent à l’état latent (cycle lysogénique). André Lwoff (Institut Pasteur, Paris), découvreur de la lysogénie, a baptisé « prophages » ces phages, dits tempérés. Esther M. Lederberg et Joshua Lederberg (Yale University) furent parmi les premiers à étudier ce phénomène (ainsi que la conjugaison, l’échange de gènes entre bactéries de la même espèce). Esther effectua l’essentiel du travail expérimental et Joshua reçut le prix Nobel de physiologie ou médecine 1958, à l’âge de 33 ans !
A l’Institut Pasteur, deux groupes de chercheurs étudiaient le phénomène « d’induction » : celui de Lwoff, avec François Jacob et Elie Wollman, s’intéressait à l’induction de phages dans des bactéries lysogènes (Pseudomona pyocanea) soumises à une irradiation par un rayonnement UV ; celui de Jacques Monod, avec l’association de Melvin Cohn, Arthur Pardee, François Gros et François Jacob, étudiait l’induction enzymatique par les β-galactosides de la β-galactosidase d’Escherichia coli. Dans les deux cas, la synthèse protéique se faisait sur les ribosomes bactériens mais, dans certaines conditions expérimentales, le phénomène d’induction était si rapide que l’on ne pouvait pas l’attribuer à une augmentation du nombre de ribosomes. Je rappelle que chez les procaryotes l’absence d’enveloppe nucléaire fait que génome et machinerie de synthèse protéique coexistent dans le même compartiment cellulaire. Entre ces deux entités, il devait exister un intermédiaire, probablement un messager de nature ribonucléotidique ; Jacob le baptisa : « intermédiaire X ». En 1960, au cours d’une réunion avec Sydney Brenner et Francis Crick, Jacob leur résuma ses résultats et son hypothèse de l’existence d’un intermédiaire instable et à vie courte. Brenner mentionna alors les résultats d’expériences sur Escherichia coli infectée par le bactériophage T2, publiés cinq ans plus tôt par Elliot Volkin et Lazarus Astrachan (Oak Ridge National Laboratory). Volkin et Astrachan avaient détecté, après l’infection, l’apparition d’une grande quantité d’ARN précédant celle d’ADN et de protéines du phage ; cet ARN avait le même rapport des proportions de bases que l’ADN du phage T2. Volkin et Astrachan lui donnèrent le nom de « DNA like RNA ». Ils pensaient avoir affaire à un précurseur de l’ADN ce qui, compte tenu de ce que l’on savait déjà du fonctionnement de l’ADN polymérase de Kornberg, était peu vraisemblable. Les résultats de Volkin et Astrachan restèrent confidentiels pendant cinq ans ! Le DNA like RNA ressemble à l’intermédiaire X instable qui apparaît lors de l’induction de la β-galactosidase. Pour tester cette hypothèse, Jacob et Brenner, dans le laboratoire de Max Delbrück (California Institute of Technology), utilisèrent la stratégie expérimentale que Meselson et Stahl avaient utilisée pour démontrer que la réplication est semi conservative. Pour Jacob et Brenner, il fallait montrer que l’intermédiaire X, apparaissant après l’infection de Coli par un phage, s’associait aux ribosomes marqués avec des isotopes lourds avant infection par le phage. Après centrifugation en gradient de densité de chlorure de césium, la fixation de l’intermédiaire X sur les ribosomes était déterminée en mesurant, dans les fractions, l’absorption UV et la radioactivité. Des expériences complémentaires pour prouver que les ribosomes ayant chargé l’intermédiaire X fabriquent les protéines du phage furent effectuées par Brenner à Cambridge. La recherche du second intermédiaire (après l’ARNt) intervenant dans la synthèse protéique avait abouti. Les articles de François Gros, Walter Gilbert, Howard Hiatt et James Watson (Harvard University), et de François Jacob, Sidney Brenner et Matthew S. Meselson (California Institute of Technology, Pasadena) furent publiés en 1961. L’ARN messager fut isolé par trois groupes de chercheurs travaillant indépendamment, dont celui de Samuel Weiss. En biologie moléculaire, la découverte de l’ARNm est l’une des plus importantes après celle de la double hélice.
Références : Lwoff A, Gutman A Recherches sur un Bacillius megaterium lysogène (1950)
Lwoff A, Siminovitch L, Kjelgaard N Induction de production de phage dans une bactérie lysogène (1950)
Lederberg EM, Lederberg J Genetic Studies of Lysogenicity in Escherichia Coli (1953)
Jacob F, Wollman EL Recherches sur les bactéries lysogènes défectives. I. Déterminisme génétique de la morphogenèse chez un bactériophage tempéré (1956)
Pardee AB, Jacob F, Monod J The genetic control and cytoplasmic expression of “Inducibility” in the synthesis of β-galactosidase by E. coli (1959)
Volkin E, Astrachan L Phosphorus incorporation in Escherichia coli ribonucleic acid after infection with bacteriophage T2 (1956)
Brenner S, Jacob F, Meselson M An unstable intermediate carrying information from genes to ribosomes for protein synthesis (1961)
Gros F, Gilbert W, Hiatt H, Kurland CG, Risebrough RW, Watson JD (1961) Unstable ribonucleic acid revealed by pulse labelling of Escherichia coli (1961)
Jacob F, Monod J (1961) Genetic regulatory mechanisms in the synthesis of proteins (1961)
« Breaking the genetic code »
Au début des années 1960, l’existence d’un code génétique apparaît comme plus que probable et l’on soupçonne qu’il est composé de triplets de bases ; reste à le démontrer. La réponse à ces interrogations fut d’abord recherchée par une approche théorique. J’ai mentionné dans le sous-chapitre précédent l’hypothèse formulée par Gamov à la suite de la publication de l’article de Watson et Crick sur la structure en double hélice de l’ADN. Gamov proposait, entre autres choses, que la sélection des acides aminés soit imposée par un code (baptisé diamond code) formé de triplets de bases nucléotidiques, une hypothèse qui s’imposa à Crick (Department of Physics, The Cavendish Laboratory, University of Cambridge). Un code constitué de triplets implique l’existence de redondances : plusieurs triplets peuvent coder pour le même acide aminé puisque le code à trois bases offre 64 possibilités pour 20 acides aminés. Francis Crick, John Griffith et le chimiste Leslie Orgel imaginèrent un modèle de « codes sans virgules » (Codes without Commas), une succession de triplets avec un seul point de départ et un seul cadre de lecture. Un tel code comprendrait 20 « triplets sens » (the magic twenties) et 44 « triplets non-sens ». Ce modèle, bien que loin de satisfaire ses auteurs, fut publié et complété par un article sur la mutagenèse. Ce fut une base de réflexion pour l’expérience qu’allait réaliser Brenner et Crick – modifier un gène du bactériophage T4 d’Escherichia coli – dans l’espoir d’aboutir à la résolution de la nature du code en évitant la laborieuse approche de la biochimie.
Références : Crick FHC, Griffith JS, Orgel LE Codes without Commas (1957)
Brenner S, Barnett L, Crick FHC, Orgel A The theory of mutagenesis (1961)
Note : L’appellation « codon » fut inventée par Sydney Brenner vers le milieu des années 1950
Sans aller dans les détails, je voudrais revenir sur l’expérience de Crick et Brenner brièvement évoquée dans le sous-chapitre précédant. Il s’agissait de répondre à la question : le code est-il constitué de triplets de bases ? L’approche expérimentale consistait à évaluer la fonctionnalité d’un gène lorsqu’on modifie son cadre de lecture en ajoutant ou supprimant une ou plusieurs bases nucléotidiques. Crick et Brenner sélectionnèrent le gène rII du bactériophage T4, qui code pour une protéine exprimée dans les premières minutes suivant l’infection d’Escherichia coli par T4. Le gène rII comprend deux cistrons contigus, A et B, codant pour les protéines rIIA et rIIB, qui sont exprimées séquentiellement. Le phage T4 présente un taux élevé de recombinaison par crossing over ; la protéine rIIB jouerait un rôle dans la recombinaison génétique. Seymour Benzer (Purdue University) a observé que dans une souche d’Escherichia coli résistante à l’infection, le gène rII de T4 était muté.
La proflavine induit des mutations en divers sites du gène rII ; ce dérivé de l’acriflavine (diamino 3,6 acridine) est utilisé en thérapeutique pour ses propriétés bactériostatiques ; c’est aussi un agent intercalant s’insérant entre deux bases de l’ADN. Le phage T4 se reproduit très rapidement dans son hôte ; lors de la réplication, la polymérase place une base en face de l’agent intercalant, introduisant ainsi une nouvelle base dans le brin néo synthétisé. E. Freeze a montré que la proflavine provoque des « mutations par transition » (une purine à la place d’une autre purine) et par « transversion » (une purine à la place d’une pyrimidine) ; Crick et Brenner montrèrent qu’elle provoque aussi des mutations par addition ou délétion de bases ; ils les ont appelées «plus frameshift mutation » (addition) et «minus frameshift mutation » (soustraction). Sur le plan expérimental, Escherichia coli, cultivé en boîtes de pétri, sur un milieu solide, donne naissance à un tapis de cellules. Le bactériophage T4 – souche « sauvage », mutée ou recombinée – est incubé dans un milieu de culture contenant Escherichia Coli ; on obtient une suspension de phages, de bactéries lysées et de quelques bactéries intactes. Une goutte de ce milieu dilué est déposée sur une plaque de pétri tapissée d’une couche de cellules bactériennes. Une bactérie infectée donne naissance à une petite colonie (une « plaque ») ; chaque plaque est issue d’une seule bactérie infectée ; son apparence est caractéristique du gène sauvage (phénotype clear plaque) ou muté (phénotype cloudy plaque dû à la présence de quelques bactéries survivantes). On peut ainsi isoler un phage muté parmi des centaines de souches sauvages. L’inverse, la reconnaissance de T4 sauvage parmi des centaines de souches mutées, se fait par le pouvoir lytique du phage sur E. coli. J’ai mentionné plus haut que le taux de recombinaison est élevé chez T4. Si on co-infecte E. coli avec deux phages mutés dans deux gènes différents, la complémentation entre les produits des deux gènes mutés (ou l’absence de complémentation) permet de reconnaître si l’on est revenu (ou pas) au type sauvage (lyse de E. coli). Crick et Brenner ont multiplié les expériences de co-infection avec des phages comportant un nombre de mutations allant de deux à six ; seules les co-infections avec trois ou six mutations ont permis de restaurer le phénotype sauvage. Le laborieux travail de Crick et Brenner a abouti à un certain nombre de conclusions et de confirmations : (i) le code génétique est formé de codons de trois bases nucléotidiques ; (ii) un triplet spécifie un acide aminé ; (iii) il existe des codons non-sens ; (iv) le code génétique est dégénéré. En établissant une cartographie détaillée (jusqu’à l’échelle du nucléotide) des mutations sur les génes rIIA et rIIB, Seymour Benzer avait montré que les codons sont alignés (la carte est linéaire) et ne se « chevauchent » pas.
Références : Benzer S Fine structure of a genetic region in bacteriophage (1955)
Brenner S, Benzer S, Barnett L Distribution of proflavin-induced mutations in the genetic fine structure (1958)
Freese E On the molecular explanation of spontaneous and induced mutations (1959)
Benzer S On the topography of the genetic fine structure (1961)
Crick FH, Barnett L, Brenner S, Watts-Tobin RJ General nature of the genetic code for proteins (1961)
Heere LJ, Karam JD Analysis of Expressions of the rII Gene Function of Bacteriophage T4 (1975)
Finalement, c’est l’approche expérimentale des biochimistes qui a permis de répondre aux deux questions sur l’existence et la nature du code. Lorsque Marshall W. Nirenberg (National Institute of Arthritic and Metabolic Diseases, Bethesda) commença à s’intéresser au mode de transmission de l’information de l’ADN aux protéines, la notion d’ARN intermédiaire était encore vague. Johann H. Matthaei prépara un système de synthèse protéique in vitro à partir d’un extrait d’Escherichia coli traité à la ribonucléase pour détruire les ARN présents dans le milieu, sauf les ARNr protégés par leur association avec les protéines ribosomiales ; l’extrait fut additionné avec le mélange des vingt acides aminés, dont la plupart étaient marqués avec un isotope radioactif. Le système fut programmé avec un ARN viral et, dans l’expérience « témoin », avec un polyuridylate (UUUUUUUUU…) obtenu en incubant la polynucléotide phosphorylase de Grunberg-Manago et Ochoa en présence d’uridine (U). Dans le modèle du code sans virgule, les triplets de trois bases identiques (AAA, GGG, CCC, UUU) étaient non-sens (codons stop). A la surprise de Matthaei, le système acellulaire témoin avait synthétisé un polypeptide constitué uniquement de phénylalanine (F-F-F…). Pour la première fois l’on établissait une correspondance entre une séquence ribonucléotidique et un acide aminé. Ce résultat, présenté en août 1961 à l’International Congress of Biochemistry, à Moscou, devant un public restreint faillit passer inaperçu ! Trois ans plus tard, Nirenberg et Philip Leder franchirent une étape supplémentaire en montrant que le système acellulaire de synthèse protéique programmé avec un polyU fixe le phenylalanyl-ARNtPhe à l’exclusion de tout autre aminoacyl-ARNt. En utilisant la même approche expérimentale, on a montré que le polycytidilate (CCCCCCCCC…) code pour la polyproline (P-P-P…) ; le polyadénylate (AAAAAA…), pour la polylysine (Y-Y-Y…).
Références : Matthaei HJ, Nirenberg MW Characteristics and Stabilization of DNAase Sensitive Protein Synthesis in E. coli Extracts (1961)
Nirenberg MW, Matthaei HJ The Dependence of Cell-Free Protein Synthesis in E. coli upon Naturally Occuring or Synthetic Polyribonucleotides (1961)
Matthaei HJ, Jones OW, Martin RG, Nirenberg MW Characteristics and composition of RNA coding units (1962)
Leder P, Nirenberg MW RNA Codewords and Protein Synthesis. The Effect of Trinucleotides upon the Binding of sRNA to Ribosomes (1964)
Leder P, Nirenberg MW RNA Codewords and Protein Synthesis, III. On the Nucleotide Sequence of a Cysteine and a Leucine RNA Codeword (1964)
L’hypothèse du triplet fut confirmée par le résultat des expériences de synthèse protéique in vitro d’Har Gobind Khorana (The University of Wisconsin, Madison, puis Massachusetts Institute of Technology). Khorana étudia la chimie dans sa province natale du Punjab, qu’il quitta lorsqu’elle passa sous juridiction pakistanaise, après la partition du sous-continent indien. Il compléta sa formation de chimiste organicien par un PhD à l’Université de Liverpool, un séjour à Zurich dans le laboratoire de Vladimir Prelog (prix Nobel de chimie, 1975) et dans celui d’Alexander R. Todd, un spécialiste de la chimie des nucléotides qui fut à deux doigts de découvrir la structure de l’ADN. Pendant le séjour de Khorana à Cambridge, Frederick Sanger achevait la séquence de l’insuline et Max Perutz, la structure tridimensionnelle de la myoglobine. Pour résoudre le problème du code, Khorana adopta d’abord la stratégie expérimentale de Marshall Nirenberg et Philip Leder ; il synthétisa des polyribonucléotides formés, non pas d’une seule base, mais d’une alternance de deux bases (UCUCUCUCU…), trois bases (AUGAUGAUG…), quatre bases… Les polyribonucléotides, amplifiés par l’ARN polymérase (Khorana inventa la Polymerase Chain Reaction avant la lettre), représentaient une quantité suffisante d’ARNm pour programmer le système acellulaire de synthèse protéique. Les polypeptides synthétisés étaient isolés et séquencés. Un problème se posa d’emblée : en utilisant plusieurs bases, leur répartition dans la séquence du polypeptide se faisait au hasard et il devenait très difficile d’établir une correspondance entre séquence nucléotidique et séquence des acides aminés. Il fallait changer de stratégie expérimentale. Khorana recourut à la chimie en milieu non-aqueux combinée à l’utilisation de polymérases et de ligases, pour synthétiser des polydésoxyribonucléotides dont la taille s’allongea jusqu’à atteindre celle de mini-gènes. En comparant leurs séquences avec celles des peptides synthétisés, Khorana confirma que le code génétique est formé de triplets (codons) ; il identifia les codons de la sérine (UCU), de la leucine (CUC) et les codons STOP qui interrompent la traduction : UAG, UAA, UGA.
En 1972, Walter Fiers et ses collaborateurs (Laboratory of Molecular Biology, Universiteit Gent, Belgique) apportèrent une preuve directe que la machinerie de transcription et de synthèse protéique lit le code génétique de manière séquentielle et par triplets. Le génome du bactériophage MS2 est un ARN. La protéine d’enveloppe. Fiers établit la séquence nucléotidique complète du gène de la protéine d’enveloppe du virus. C’est le premier gène dont on ait établi la séquence. Il la compara à la séquence des 128 résidus d’acides aminés de la protéine d’enveloppe. Les deux séquences s’alignaient parfaitement ; chaque codon du gène était disposé dans le même ordre que l’acide aminé correspondant. En 1976, Khorana parvint à un résultat similaire après avoir synthétisé un gène et comparé sa séquence à celle du polypeptide obtenu par transcription et traduction in vitro. Le déchiffrage complet des 64 triplets du code génétique fut achevé en 1966. En 1968, Khorana partagea le prix Nobel de physiologie ou médecine avec Robert W. Holley et Marshall Nirenberg. En 1976, Fiers élucida la première séquence complète d’un génome viral, celui du bactériophage MS2.
Références : Khorana HG, Büchi H, Ghosh H, Gupta N, Jacob TM, Kossel H, Morgan R, Narang SA, Ohtsuka E, Wells RD Polynucleotide synthesis and the genetic code (1966)
Min Jou W, Haegeman G, Ysebaert M, Fiers W Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein (1972)
Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, Min Jou W, Molemans F, Raeymaekers A, Van den Berghe A, Volckaert G, Ysebaert M Complete nucleotide-sequence of bacteriophage MS2-RNA – primary and secondary structure of the replicase gene (1976)
ARN polymérases
L’âpre compétition entre laboratoires pour découvrir l’ARN polymérase ADN-dépendante a été relancée lorsqu’il fut établi que la polynucléotide phosphorylase de Marianne Grunberg Manago et Severo Ochoa n’est pas ADN-dépendante – dans la cellule, elle fonctionne comme une ribonucléase ! Parmi les vainqueurs de la compétition, dont les articles furent publiés simultanément en 1960, figure Audrey Stevens, une étudiante postdoctorale qui faillit coiffer sur le poteau tous les groupes lancés dans la quête de l’ARN polymérase ADN-dépendante.
En 1959, Samuel B. Weiss (Department of Biochemistry, University of Chicago) mit en évidence dans une fraction nucléaire de foie de rat un enzyme catalysant l’incorporation de P32CTP dans de l’ARN en présence d’ATP, UTP, GTP ou CTP. L’absence de l’un des quatre ribonucléosides triphosphates dans le milieu d’incubation diminuait le taux d’incorporation ; par contre le taux d’incorporation était peu sensible à un traitement à la désoxyribonucléase. Le produit formé était sensible à la RNAse et libérait du P32CTP par hydrolyse alcaline. En 1960, Weiss et Tokimasa Nakamoto (Argonne Cancer Research Hospital) isolèrent l’ARN polymérase ADN-dépendante de la bactérie Micrococcus lysodeikticus. L’ARN synthétisé avait la même composition nucléotidique que l’ADN ayant servi de matrice. La même année, Jerard Hurwitz (New York University School of Medicine) isola, à partir d’Escherichia Coli, un enzyme répondant à tous les critères requis : synthèse d’ARN seulement en présence des quatre nucléotides ; stimulation du taux d’incorporation des nucléotides marqués après addition d’ADN de différentes sources ; inhibition par l’addition de ADNase ou de ARNase. En 1960, parut un article d’Audrey Stevens qui, dans le laboratoire de Léon A. Heppel (National Institute of Health, Bethesda), rapportait la présence, dans des extraits d’Escherichia Coli, d’un enzyme incorporant P32ATP dans de l’ARN en présence des quatre nucléotides ; l’activité détectée était sensible à la RNAse. Ru-Chih C. Huang, dans le laboratoire de James Bonner (California Institute of Technology), caractérisa, dans des extraits d’embryons de pois, la première ARN-polymérase chez les plantes.
Références : Weiss SB, Gladstone L A Mammalian System for the Incorporation of Cytidine Triphosphate into Ribonucleic Acid (1959)
Weiss SB, Nakamoto T Net Synthesis of Ribonucleic Acid with a Microbial Enzyme Requiring Deoxyribonucleic Acid and Four Ribonucleosides Triphosphates (1960)
Hurwitz J, Bressler A, Diriger R The enzymic incorporation of ribonucleotides into polyribonucleotides and the effect of DNA (1960)
Stevens A Incorporation of the adenine ribonucleotide into RNA by cell fractions from E. coli B (1960)
Huang RC, Maheshwari N, Bonner J Enzymatic synthesis of RNA (1960)
Les cellules procaryotes n’ont qu’une ARN polymérase ADN-dépendante; les cellules eucaryotes ont en trois : l’ARN polymérase I transcrit l’ARN les gènes des précurseurs des ARNr 28S, 18S, 5,8S ; l’ARN polymérase II transcrit les gènes des ARNm ; l’ARN polymérase III transcrit les gènes de l’ARN de transfert (ARNt), des petits ARN : ARNsn (small nuclear), ARNsi (small interfering), microARN (ARNmi), ARNr 5S, ARN 7S, et les ARN du splicéosome : ARNsn U1 à ARNsn U6, ARN de voûte (ARNv) associé aux pores nucléaires… L’ARN polymérase ADN-dépendante est un complexe enzymatique dont l’activité – la « transcription » selon le terme proposé par Sol Spiegelman et Masaki Hayashi – est hautement régulée par des facteurs de transcription. En 2003, David A. Bushnell et Roger D. Kornberg (Stanford University), le fils d’Arthur Kornberg, résolurent la structure cristallographique et le mécanisme d’action à l’échelle atomique de l’ARN polymérase II de Saccharomyces cerevisiae. Kornberg reçut le prix Nobel de chimie 2006.
Référence : Bushnell DA, Kornberg RD Complete RNA polymerase II at 4.1 Å resolution: implications for the initiation of transcription (2003)
Enzymes de restriction
Au début des années 1950, Salvador Luria et son étudiante, Mary Human (Indiana University, Bloomington), comparaient l’interaction de bactériophages avec différentes souches d’Escherichia coli. Ils observèrent que les bactériophages ayant infecté certaines souches avaient subi des modifications – non-héréditaires – qu’ils baptisèrent « modification induite par l’hôte » (host-induced modification) et qui affectent la capacité des bactériophages d’infecter d’autres souches de E. coli ou la bactérie : Shigella dysenteriae.
Référence : Luria S, Human M A nonhereditary, host-induced variation of bacterial viruses (1952)
Lorsqu’un bactériophage envahit une bactérie il peut, selon les circonstances, opter pour deux cycles de vie : la multiplication immédiate avec lyse de la bactérie (cycle lytique), ou l’intégration de son génome dans le chromosome bactérien – ou dans un plasmide. Le phage intégré (prophage) se réplique en même temps que la bactérie. Dans le laboratoire de Salvador Luria, Giuseppe Bertani étudiait la lysogénie sur une souche d’Escherichia coli (Lisbonne strain) et une souche de Shigella. Il montra que la production de phages, chez les bactéries lysogènes, est limitée à quelques rares épisodes. Qu’est-ce qui limite ? Parmi les trois phages – P1, P2, P3 – qui sont libérés, Bertani concentra son étude sur le phage non inductible P2. Avec Jean J. Weigle, ils montrèrent qu’à côté du phénomène de lysogénie coexiste un autre phénomène qu’ils appelèrent « variation contrôlée par l’hôte » (host controlled variation). Ce n’est qu’avec la découverte des endonucléases de restriction, quinze ans plus tard, que l’on comprendra la signification et la portée de ce phénomène.
Références : Bertani G Studies on lysogenesis. 1. The mode of phage liberation by lysogenic Escherichia coli (1951)
Bertani G, Weigle JJ Host controlled variation in bacterial viruses (1953)
Après sa thèse de doctorat sur les mutants défectifs du bactériophage λ (1958), au California Institute of Technology, sous la direction de Jean J. Weigle, un disciple de Salvador Luria, Werner Arber poursuivit ses travaux dans son institution d’origine (Institut de Biophysique, Faculté des Sciences, Genève). En virologie, le terme « restriction » fait allusion à la restriction à la règle de sensibilité des bactéries aux bactériophages. Au cours de son travail de thèse sur l’infection par le bactériophage λ, Daisy Dussoix a découvert le système de « restriction-modification de l’ADN » (host-controlled modification) qui protège les bactéries contre les infections virales. Les résultats des travaux qu’elle a effectués avec Arber au début des années 1960 ont montré que le phénomène de restriction consiste à induire des changements dans l’ADN du phage, aboutissant à sa dégradation. Les bactéries possèdent donc la capacité de contrecarrer l’infection par un phage en détruisant son ADN, même si certains phages continuent à être exprimés dans la bactérie. Il restait à apporter la preuve expérimentale que la bactérie a la capacité de modifier l’ADN viral. Arber avança la théorie que chaque souche bactérienne synthétise une endonucléase capable de reconnaître une séquence spécifique de nucléotides dans un ADN étranger, et de l’exciser. Si chaque souche bactérienne possède des nucléases, comment protège-t-elle son ADN contre l’action de ces enzymes ? Je rappelle que chez les procaryotes l’ADN chromosomique n’est pas protégé par une enveloppe nucléaire, comme dans les cellules eucaryotes. En 1968, Stuart Lin et Arber ont décrit la présence, dans une souche d’Escherichia coli, d’un enzyme dont l’activité requiert la présence de S-adénosyl-méthionine, un coenzyme donneur de groupes méthyl ; une incubation de l’ADN de la forme réplicative du phage fd avec cet enzyme confère à l’ADN viral une résistance à la restriction. La protection de l’ADN bactérien est assurée par des méthylases, qui ajoutent des groupes méthyl sur des sites spécifiques de l’ADN.
Références : Arber W, Dussoix D Host specificity of DNA produced by Escherichia coli. I. Host controlled modification of bacteriophage lambda (1962)
Dussoix D, Arber W Host specificity of DNA produced by Escherichia coli. II. Control over acceptance of DNA from infecting phage lambda (1962)
Arber W, Hattman S, Dussoix D On the Host-Controlled Modification of Bacteriophage Lambda (1963)
Linn S, Arber W Host specificity of DNA produced by Escherichia coli, X. In vivo restriction of phage fd replicative form (1968)
En résumé, le système de restriction-modification permet à Escherichia coli de modifier son ADN ou celui des phages. L’addition de groupes chimiques induit une résistance aux enzymes de restriction. Introduites dans l’ADN d’un phage, ces modifications affectent la capacité du virus à être accepté ou rejeté par un nouvel hôte. L’absence de modification permet à la bactérie de détecter la présence d’un ADN étranger, ce qui désigne ce dernier à la dégradation. Pour un phage donné, la spécificité de l’hôte est encodée dans son ADN ; la bactérie hôte laisse une « signature » dans l’ADN du phage qui l’a infectée.
Référence : Arber W Promotion and limitation of genetic exchange (1979)
La caractérisation des endonucléases de restriction fut laborieuse ; à la fin des années 1960, on n’en connaissait que quatre. Les enzymes de Type I reconnaissent un site de restriction spécifique sur un ADN, mais ils le fragmentent au hasard, à proximité ou à distance du site de reconnaissance, ce qui rend impossible leur utilisation comme outils pour les manipulations génétiques. Le premier enzyme de restriction de Type II – reconnaissant un site de restriction spécifique et coupant les deux brins d’ADN exclusivement au niveau de ce site – fut isolé par Hamilton O. Smith et coll. (Department of Microbiology, John Hopkins University School of Medicine) à partir d’Haemophilus influenzae infecté par le bactériophage P22.
Références : Smith HO, Welcox KW A Restriction enzyme from Hemophilus influenzae: I. Purification and general properties (1970)
Kelly TJ, Smith HO A restriction endonuclease from Hemophilus influenzae. II. Base sequence of the recognition site (1970)
Cette découverte incita Daniels Nathans (Department of Microbiology, John Hopkins University School of Medicine, Baltimore) à envisager l’utilisation d’enzymes de restriction pour disséquer le génome de SV40 (Simian Virus 40), un virus oncogène à ADN. Il demanda à son étudiant, Stuart Adler, de tester les quatre endonucléases connues sur cet ADN. Seule l’endonucléase découverte par Smith coupe cet ADN en fragments spécifiques. Nathan, et son étudiante graduée Kathleen J. Danna, séparèrent les 11 fragments par électrophorèse en gel de polyacrylamide et établirent leur séquence. En se basant sur les chevauchements de séquences entre fragments, ils les alignèrent et établirent une ébauche de carte génomique de l’ADN de SV 40. La découverte de nouveaux enzymes de restriction de type II permit à Nathan, Danna Adler, George Sack, et les autres membres du groupe de compléter cette première carte du génomique, avec la position des sites de clivage. La collaboration du groupe de Nathans avec George Khoury et Malcolm Martin (Laboratory of Biology of Viruses National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda) aboutit à l’établissement d’une carte des ARNm du virus SV 40 et à la localisation d’un certain nombre de gènes de structure : pour la première fois, on disposait d’une carte génomique d’un virus à ADN. Werner Arber, Daniels Nathans et Hamilton Smith partagèrent le prix Nobel de Médecine ou Physiologie 1978, pour la découverte des enzymes de restriction et leur application aux problèmes de génétique moléculaire.
Références : Danna KJ, Nathans D Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae (1971)
Danna KJ, Sack GH, Nathans D Studies of Simian virus 40 DNA: VII. A cleavage map of the SV40 genome (1973)
Khoury G, Martin MA, Lee TNH, Danna KJ, Nathans D A map of simian virus 40 transcription sites expressed in productively infected cells (1973)
Les généticiens moléculaires disposent, aujourd’hui, de plusieurs centaines d’enzymes de restriction. Ils sont nommés d’après leur origine : BamHI (Bacillus amyloliquefaciens), EcoRI (Escherichia coli), MspI (Moraxella catarrhalis), Sau3A (Staphylococcus aureus 3A), KpnI (Klebsiella pneumoniae)… Les sites de restriction sont souvent palindromiques ; les séquences nucléotidiques identiques ont des orientations antiparallèles sur les deux brins ; par exemple, pour BamHI :
GGATCC
CCTAGG
Après coupure de l’ADN, les deux brins situés aux extrémités des fragments peuvent être des bouts francs (blunt ends) de longueur égale, ou des bouts collants (sticky ends) de longueur inégale, ce qui facilite la recombinaison des fragments isolés. Au cours des années 1980, de nouvelles perspectives dans le domaine du clonage se sont ouvertes lorsqu’on a découvert que les ADN d’origine végétale et animal sont des substrats pour les enzymes de restriction.
Une nouvelle classe d’enzymes de restriction, baptisée Type IV, a fait son apparition dans les années 1990. A la différence des enzymes des classes I, II, et III, ils ne reconnaissent pas une séquence spécifique de l’ADN mais des sites portant des bases nucléotidiques méthylées : cytosine pour McrB et McrC (Modified cytosine restriction) et adénine pour Mrr (Modified DNA rejection and restriction) d’Escherichia coli ; cette modification doit être située à l’endroit ou à proximité du site de clivage. Ces endonucléases sont constituées de deux domaines distincts (ou deux sous-unités), l’une pour la reconnaissance du site et l’autre pour la coupure de l’ADN bi-caténaire. Les enzymes de type IV ne coupent pas l’ADN non-méthylé ; ils n’ont pas de méthyltransférases correspondantes. Leur fonction principale est la protection d’Escherichia coli contre les bactériophages dont l’ADN a été méthylé par d’autres bactéries, et la régulations des modifications épigénétiques qui affectent l’expression des gènes sans en modifier la séquence nucléotidique.
Références : Waite-Rees PA, Keating CJ, Moran LS, Slatko BE, Hornstra LJ, Benner JS Characterization and expression of the Escherichia coli Mrr restriction system (1991)
Janulaitis A, Vaisvila R, Timinskas A, Klimasauskas S, Butkus V Cloning and sequence analysis of the genes coding for Eco57I restriction-modification enzymes (1992)
Des efforts ont été déployés pour découvrir des ciseaux moléculaires ayant de nouvelles spécificités de reconnaissance. Trois approches ont été utilisées : (i) le criblages de bactéries et de micro-organismes – un procédé long et fastidieux ; (ii) l’introduction de mutations dans la séquence d’endonucléases de restriction existantes ; (iii) la synthèse de nouveaux enzymes en fusionnant des structures en doigts de zinc avec le domaine catalytique d’une endonucléase. Les doigts de zinc sont des petites structures tridimensionnelles, souvent présentes dans les zones d’interaction entre facteurs de transcription et ADN. Dans le doigt de zinc Cys2His2, un ion Zn2+ est complexé entre deux histidines d’une hélice α et deux cystéines d’un feuillet β, antiparallèles. La zone d’interaction entre la partie C-terminale d’un doigt de zinc et l’ADN est relativement courte : environ 3 paires de bases ; on peut la porter à une vingtaine de paires de bases en construisant une chimère associant à la nucléase plusieurs motifs doigts de zinc (6 à 8). Les nucléases à doigts de zinc présentent l’avantage relatif d’être de petite taille ; elles présentent l’inconvénient d’être moins précises.
En sa qualité de spécialiste de la synthèse de macromolécules biologiques, Srinivasan Chandrasegaran a participé à l’entreprise internationale ayant abouti à la synthèse du premier chromosome fonctionnel de levure. Avec ses collègues du Department of Environmental Health Sciences (Johns Hopkins University, School of Hygiene and Public Health, Baltimore), ils ont associé au domaine catalytique de l’endonucléase de restriction FokI, de Flavobacterium okeanokoites, deux protéines comportant chacune trois doigts de zinc. Bien que FokI soit un enzyme monomérique, c’est seulement en recrutant un deuxième monomère, lié ou non à l’ADN, que FokI coupe les deux brins d’ADN en deux endroits distincts, à une certaine distance de la séquence cible. La liaison des deux protéines à doigts de zinc à leurs séquences respectives d’ADN, éloignées de quelques nucléotides, rapproche les deux monomères pour former le dimère actif qui clive l’ADN.
Référence : Kim YG, Cha J, Chandrasegaran S Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain (1996)
Ingénierie génétique moléculaire
Manipuler le génome. Les premières expériences de transgenèse remontent au début des années 1970 : Paul Berg et coll. (Department of Biochemistry, Stanford University Medical Center, prix Nobel de chimie 1980) ont inséré dans le génome d’Escherichia coli un fragment d’ADN du virus SV40. Herbert W. Boyer (Department of Biochemistry and Biophysics, University of California, San Francisco) perfectionna la technique en utilisant comme vecteur un petit ADN circulaire. Pour la première fois, on était parvenu à introduire un gène étranger dans un génome bactérien, mais on n’était pas en mesure de prédire le site d’insertion.
Références : Jackson DA, Symons RH, Berg P Biochemical Method for Inserting New Genetic Information into DNA of Simian Virus 40: Circular SV40 DNA Molecules Containing Lambda Phage Genes and the Galactose Operon of Escherichia coli (1972)
Itakura, K; Hirose, T; Crea, R, Riggs AD, Heyneker HL, Bolivar F, Boyer HW Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin (1977)
Dans les années 1980, Renato Dulbecco (Department of Bacteriology, Indiana University) et Albert Kelner (The Biological Laboratory, Cold Spring Harbor, New York), les découvreurs des mécanismes de réparation de l’ADN, eurent recours à la recombinaison homologue ; cette voie, très conservée pour maintenir la stabilité génétique des espèces, permet de corriger des mutations pathogènes en insérant des gènes dans le génome de cellules vivantes, ou en les modifiant (gene targeting). Le fragment de chromosome portant la mutation est remplacé par une séquence homologue définie par l’expérimentateur. Pour rappel, on désigne par les termes « recombinaison homologue » l’échange de matériel génétique entre deux molécules d’ADN homologues. Ce processus intervient dans la réparation des cassures double brin de l’ADN ; un ADN homologue sert de matrice. Cette approche expérimentale permet d’éviter l’incertitude concernant la localisation du gène corrigé.
Références : Dulbecco R Reactivation of ultra-violet-inactivated bacteriophage by visible light (1949)
Kelner A Effect of Visible Light on the Recovery of Streptomyces Griseus Conidia from Ultra-violet Irradiation Injury (1949)
Dans les années 2000, sont apparues les nucléases à doigts de Zn, puis la technologie TALEN. Les nucléases à doigts de Zn permettent la modification ciblée de l’ADN par l’insertion, la suppression ou le remplacement de segments d’ADN au site de coupure double brin de l’ADN. La technique TALEN est également basée sur l’utilisation de nucléases programmées pour trouver, avec une bonne précision, des séquences cibles d’ADN, effectuer une coupure double brin et déclencher un mécanisme de réparation cellulaire. En reconnaissant une séquence particulière d’ADN, la protéine TAL sert de guide à la nucléase qui lui est associée.
Dans la mutagénèse dirigée par oligonucléotide, il n’y a pas de coupure d’ADN ; on utilise des segments synthétiques d’ADN ou des oligonucléotides hybrides ADN/ARN. Ce sont de courtes séquences d’ADN synthétisées en laboratoire puis amplifiées par réaction en chaîne par polymérase (PCR). Le fragment d’ADN, complémentaire de la séquence d’ADN du gène cible à modifier, est introduit dans la cellule bactérienne ou eucaryote par un vecteur (plasmide) ; sa séquence est presque identique à celle de l’ADN cible, à l’exception de quelques nucléotides (1 à 4). L’ADN polymérase du système de réplication de la cellule reproduira la nouvelle séquence du gène. La mutagénèse dirigée par oligonucléotide est utilisée pour introduire des substitutions, insertions ou délétions dans l’ADN du gène cible.
En 2012, l’introduction du système CRISPR/Cas9 a révolutionné le domaine de l’édition génomique et rendu caduques les techniques plus anciennes.
Présence de séquences répétitives dans les génomes bactériens. Chez Escherichia coli, la phosphatase alcaline existe sous trois isoformes ; la conversion post-traductionnelle de l’isozyme 1 en isozymes 2 et 3 est catalysée par le produit du gène iap, une protéine membranaire (Iap) formée de 345 acides aminés, avec une séquence signal amino-terminale de 24 résidus. Le séquençage par Yoshizumi Ishino et coll. (Department of Experimental Chemotherapy, The Research Institute for Microbial Diseases, Osaka University) du gène iap et des régions non-codantes qui le flanquent a révélé la présence, en amont du gène, d’une « unusual structure… Five highly homologous sequences of 29 nucleotides were arranged as direct repeats with 32 nucleotides as spacing ».
Référence : Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product (1987)
De courtes répétitions palindromiques groupées et régulièrement espacées furent observées dans d’autres lignées cellulaires, chez des Archées (Haloferax mediterranei, Haloferax volcanii) et des bactéries (Streptococcus pyogenes, Escherichia coli, Thermotoga maritima, Mycobacterium tuberculosis). Elles furent désignées différemment selon les auteurs : Tandem Repeats in Eukaryotic Proteins, Short Regularly Spaced Repeats (Francisco M. Mojica et coll. Departamento de Genética y Microbiología, Universidad de Alicante) ; Spacer Interspaced Direct Repeats (Ruud Jansen et coll. Department of Infectious Diseases and Immunology, Veterinary Faculty, Utrecht University); Direct Variable Repeats Spacers (Alicia Aranaz et coll. Departamento de Sanidad Animal, Facultad de Veterinaria, Universidad Complutense de Madrid). Tous ces groupes de séquences apparurent comme des variations autour d’une même structure ; pour en clarifier la nomenclature, Juan F.M. Mojica et Ruud Jansen proposèrent de les désigner sous l’appellation commune de « Clustered Regularly Interspaced Short Palindromic Repeats », acronyme : CRISPR.
Références : Mojica FMJ, Ferrer C, Juez G, Rodríguez-Valera F Long stretches of short tandem repeats are present in the largest replicons of the Archæa Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning (1995)
Mojica F, Diez-Villaseñor C, Soria E, Juez G Biological significance of a family of regularly spaced repeats in the genomes of Archæa, Bacteria and mitochondria (2000)
Jansen R, van Embden JDA, Gaastra W, Schouls LM Identification of a novel family of sequence repeats among prokaryotes (2002)
Jansen R, van Embden JDA, Gaastra W, Schouls LM Identification of genes that are associated with DNA repeats in prokaryotes (2002)
Aranaz A, Romero B, Montero N, Alvarez J, Bezos J, de Juan L, Mateos A, Dominguez L Spoligotyping profile change caused by deletion of a direct variable repeat in a Mycobacterium tuberculosis isogenic laboratory strain (2004)
Les séquences répétitives sont impliquées dans le transfert latéral de gènes. La position dans le génome de loci renfermant des séquences répétitives (regularly spaced short repeats) a été déterminée par le séquençage du génome d’une Archaea : Methanococcus jannaschii (John Craig Venter et les biologistes moléculaires de Celera Genomics, Rockville, Maryland) et celui du génome d’une bactérie : Thermotoga maritima (Paul Blum et coll. School of Biological Sciences, University of Nebraska, Lincoln). Les séquences répétitives chez les Archaea sont situées à proximité de groupes de gènes présentant des similitudes avec des gènes bactériens.
Références : Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, Kerlavage AR, Dougherty BA, Tomb JF, Adams MD, Reich CI, Overbeek R, Kirkness EF, Weinstock KG, Merrick JM, Glodek A, Scott JL, Geoghegan NS, Venter JC Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii (1996)
Singh R, Gradnigo J, White D, Lipzen A, Martin J, Schackwitz, Moriyama E, Blum P Complete Genome Sequence of an Evolved Thermotoga maritima Isolate (2015)
L’hypothèse d’un transfert d’ADN associé à des séquences répétitives entre Archaea et bactéries a été accrédité par un certain nombre d’observations expérimentales. La présence d’ADN d’Archaea dans un génome est repérable. Si le génome des Archaea est un chromosomecirculaire – comme celui des bactéries, la machinerie génétique (histones, enzymes de réplication, transcription, traduction – est proche de celle des eucaryotes. De plus, le génome des Archaea renferme des gènes particuliers codant pour des protéines permettant la vie dans des conditions extrêmes. Thermotoga maritima est une bactérie hyperthermophile vivant dans les sources chaudes, terrestres et sous-marines, à des températures de 55 à 90°C ; son génome de Thermotoga maritima est composé de 1.860.725 pb ; environ la moitié de ce génome est représenté par 1.877 régions codantes putatives ; la comparaison avec le génome d’Archaea a révélé la présence de nombreuses similarités. Depuis la fin du XXe siècle, la mise à la disposition des chercheurs d’un nombre croissant de séquences génomiques a rendu possible les travaux de génomique comparative. En comparant les génomes d’Archaea et de bactéries thermophiles, Claire M. Fraser et coll. (Institute for Genomic Research, Rockville, Maryland) ont confirmé l’existence de similitudes entre une centaine de gènes de Thermotoga maritima et ceux d’Archaea et d’autres bactéries thermophiles (108 gènes orthologues). Dans la moitié du génome de Thermotoga maritima, on observe la présence de gènes groupés dans 15 régions de 4 to 20 kilobases, disposées dans le même ordre que dans le génome d’Archaea. Ces observations sont en accord avec l’hypothèse d’un transfert latéral de gènes d’Archaea à Thermotoga maritima, impliquant les Long Clusters of Tandem Repeats présents chez les Archaea. L’évolution se fait donc par deux mécanismes : le transfert vertical de gènes des parents à leur descendance et le transfert latéral de gènes bactériens.
Références : Nelson KE, Clayton, RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White O, Salzberg L, Smith HO, Venter JC, Fraser CM Evidence for lateral gene transfer between Archæa and bacteria from genome sequence of Thermotoga maritima (1999)
Nesbo CL, L’Haridon S, Stetter KO, Doolittle WF Phylogenetic analyses ot two “archeal” genes in thermogata maritima reveal multiple transfers between archea and bacteria (2001)
Comment les bactéries hyperthermophiles protègent leur génome contre les risques de mutations dues aux températures extrêmes. Les Archaea hyperthermophiles et les bactéries Thermotoga maritima, Aquifex aeolicus, Pyrococcus furiosus, Strain 121 parviennent à survivre à des températures – jusqu’à 100°C – où les deux brins d’ADN ont tendance à se se séparer et où les bases subissent des dommages irréversibles (dépurination, désamination). Pour maintenir leur intégrité génomique en milieux hostiles, ces bactéries ont développé des mécanismes efficaces pour empêcher la dénaturation de leur ADN et connaître un taux de mutation de valeur similaire à celui des bactéries mésophiles : (i) Elles possèdent une topo-isomérase de type I, la gyrase réverse, qui introduit des supertours positifs dans l’ADN, ce qui augmente notablement sa température de fusion. (ii) Leur ADN est revêtu de petites protéines basiques insérées dans le petit sillon de manière à empêcher l’hydrolyse thermique ; chez l’archée Sulfolobus acidocaldarius, découverte dans les geysers de Yellowstone, la protéine Sac7d (7 kDa) protège l’ADN jusqu’à 85°C. (iii) Elles possèdent la boite à outils « Base Excision Repair », composée d’une glycosylase, une endonucléase, une polymérase et une ligase, pour réparer les dommages infligés aux bases – oxydation, désamination, alkylation -, ainsi qu’un mécanisme d’une grande rapidité et fidélité pour réparer, par recombinaison, les cassures double-brin de l’ADN.
Kira S. Makarova et coll. (National Center for Biotechnology Information, National Institutes of Health, Bethesda) a identifié chez les organismes thermophiles les composants d’un système de réparation de l’ADN d’un type nouveau, en recherchant des régions conservées dans les génomes de procaryotes ; un groupe d’une vingtaine de gènes répondait à ce critère. L’association de ces gènes en complexe suggérait une fonction commune ; au centre, un groupe de gènes étaient particulièrement bien conservés ; les résultats de l’analyse des séquences prédisaient la présence d’une ADN hélicase, d’une hydrolase de la superfamille HD (hydrolyse des liaisons phosphodiester), d’une exonucléase de la famille RecB (réparation des cassures double brin de l’ADN), d’une autre nucléase d’un type nouveau ; au voisinage de ce groupe, se trouvaient encore, de façon assez constante, une hydrolase de la superfamille HD (une autre nucléase ?), une ADN polymérase d’un type nouveau et une superfamille de protéines issues de gènes dupliqués (paralogues). Fonctionnellement, ce nouveau système de réparation de l’ADN présente des analogies avec le système de réparation trans lésionnel de l’ADN présent chez les bactéries et les eucaryotes. Ce mécanisme met en jeu des polymérases de la famille Y qui permettent de poursuivre la réplication d’un ADN en insérant des nucléotides en face de bases endommagées.
Référence : Makarova KS, Aravind L, Grishin NV, Rogozin IB, Koonin EV A DNA repair system specific for thermophilic archæa and bacteria predicted by genomic context analysis (2002)
Articles de revue : Van der Oost J, Ciaramella M, Moracci M, Pisani FM, Rossi M, de Vos WM Molecular biology of hyperthermophilic Archaea (1998)
López-García P DNA supercoiling and temperature adaptation: A clue to early diversification of life? (1999)
Grogan DW Stability and repair of DNA in hyperthermophilic Archaea (2004)
Comment les bactéries protègent leur génome contre l’attaque par les bactériophages. La membrane plasmique des bactéries est une bicouche phospholipidique entourée d’une (bactérie gram–) ou de plusieurs (bactérie gram+) couches de peptidoglycanes et d’une paroi de muréine (bactérie gram–) ; pour pénétrer dans la cellule les phages utilisent comme porte d’entrée les glycoprotéines intégrales de la membrane plasmique et de la paroi. Les bactéries, et la plupart des Archaea se sont dotées d’une panoplie de stratégies de défense contre les éléments génétiques invasifs (phages, virus, plasmides).
Articles de revue : Labrie SJ, Samson JE, Moineau S Bacteriophage resistance mechanisms (2010)
Hampton HG, Watson BN, Fineran PC The arms race between bacteria and their phages (2020)
(i) Les bactéries modifient la structure ou la composition en sucres des récepteurs de surface pour empêcher la fixation du phage. (ii) Elles attaquent l’ADN exogène en mobilisant leurs endonucléases de restriction-modification ; l’ADN bactérien est protégé contre ces endonucléases par méthylation de bases. (iii) Les bactéries mettent en œuvre les prophages du système de Superinfection exclusion (Sie) pour bloquer l’injection d’ADN viral à travers la membrane cellulaire.
Référence : Bucher MJ, Czyz DM Phage against the Machine: The SIE-ence of Superinfection Exclusion (2024)
(iv) Les bactéries développent une immunité adaptative. Le génome bactérien contient des locis constitués de courtes répétitions palindromiques groupées et régulièrement espacées (Clustered regularly interspaced short palindromic repeats, CRISPR). Les bactéries intègrent dans leur locus CRISPR de courtes séquences de l’ADN du phage appelées « espaceurs ». Les locis CRISPR sont des structures dynamiques archivant continuellement de nouveaux espaceurs. Hélène Deveau et coll. (Département de Biochimie et de Microbiologie, Félix d’Hérelle Reference Center for Bacterial Viruses, Université Laval, Québec), en collaboration avec Rodolphe Barrangou (Danisco USA Inc., Madison), ont montré que l’efficacité du mécanisme de résistance CRISPR1 aux deux principaux groupes de phages de Streptococcus thermophilus était en rapport avec l’insertion d’un nouvel espaceur d’une trentaine de nucléotides dans CRISPR1. En comparant la séquence de l’espaceur à celle du génome du phage, ils ont repéré la région (proto-espaceur) qui confère le phénotype « résistance au phage » à la bactérie. En intégrant de manière itérative dans le locus CRISPR de nouveaux marqueurs immunitaires provenant d’agents exogènes, les bactéries se constituent une « mémoire génétique de vaccination », dont les éléments sont transcrits et modifiés, lors d’une nouvelle infection, pour produire des ARN guides (small interfering RNAs) ; ces ARN dirigent les nucléases qui vont couper spécifiquement les séquencescomplémentaires de l’ADN viral.
Références : Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P CRISPR provides acquired resistance against viruses in prokaryotes (2007)
Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau SJ Phage response to CRISPR-encoded resistance in Streptococcus thermophilus (2008)
Barrangou R, Marraffini LA CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity (2014)
Au XXe siècle, deux découvertes ont bouleversé les connaissances dans le domaine des ARN : (i) la capacité catalytique de certains d’entre eux, mise en évidence par Thomas Cech (University of Colorado, Boulder) à la fin des années 1980 ; (ii) l’existence de petits ARN non codants (∼20−30 nucléotides). Chez Caenorhabditis elegans, à la fin du premier stade du développement larvaire, l’expression de la protéine LIN-14 est régulée négativement par le produit du gène lin-4. En 1993, Victor Ambros et coll. (Harvard University, Department of Cellular and Developmental Biology, Cambridge, Massachusetts) et Gary Ruvkun (Department of Molecular Biology, Massachusetts General Hospital, Boston) ont montré que ce produit est un petit ARN et qu’il inhibe la synthèse de LIN-14 en se fixant sur son ARNm. En 2001, let-7, un autre petit ARN impliqué dans le contrôle des stades du développement de Caenorhabditis elegans, a été découvert par les chercheurs du groupe de Thomas Tuschl (Department of Cellular Biochemistry, Max Planck Institute for Biophysical Chemistry, Göttingen). David P. Bartel et coll. (Whitehead Institute for Biomedical Research et Department of Biology, Massachusetts Institute of Technology, Cambridge) ont montré que les Small Temporal RNA (stRNAs) lin-4 et let-7 appartiennent à une classe de petits ARN non-codants, de 21 à 24-nucléotides, appelés micro-ARNs (miARN); ils en ont répertorié 55 nouveaux chez Caenorhabditis elegans. Leur importance dans la régulation de l’expression des gènes est soulignée par le fait qu’ils sont très conservés à travers les espèces. Leur fonction est la répression de la traduction de certains ARNm ou leur dégradation (gene silencing). Thomas Cech a reçu le Prix Nobel de chimie 1989 ;Victor Ambros a partagé avec Gary Ruvkun le prix Nobel de physiologie ou médecine 2024.
Références : Lee RC, Feinbaum RL, Ambros V The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14 (1993)
Wightman B, Ha I, Ruvkun G Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans (1993)
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T Identification of novel genes coding for small expressed RNAs (2001)
Lau NC, Lim LP, Weinstein EG, Bartel DP An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans (2001)
Lee RC, Ambros V An extensive class of small RNAs in Caenorhabditis elegans (2001)
Meister G, Tuschl T Mechanisms of gene silencing by double-stranded RNA (2004)
ARN transactivateur et maturation des ARN CRISPR. A l’époque où Emmanuelle Charpentier et ses étudiants, Elitza Deltcheva et Krzysztof J. Chylinski, entreprirent leurs travaux, le mécanisme de fonctionnement du système CRISPR/Cas (Clustered, Regularly Interspaced Short PalindromicRepeats/CRISPR-associated proteins) tout en suscitant un intérêt considérable de la part des chercheurs était loin d’avoir été élucidé. On savait que le processus qui permet au système immunitaire adaptatif des bactéries et des Archaea de les protéger contre les bactériophages et les plasmides, est basé sur l’intégration dans le locus CRISPR d’espaceurs d’ADN viral ou de plasmide, séparés par des séquences répétitives : « séquences répétées – espaceurs – séquence répétées – espaceurs….. ». Le mécanisme par lequel la transcription des séquences du locus CRISP produit le précurseur d’ARNcr (preARNcr), à partir duquel sont générées des unités d’espaceur répétitives uniques : les ARN CRISPR (ARNcr) restait à définir. Elitza Deltcheva et coll. (The Laboratory for Molecular Infection Medicine Sweden, Umeå Centre for Microbial Research, Department of Molecular Biology, Umeå University, Sweden) ont découvert, par criblage différentiel informatique du génome de Streptococcus pyogenes, un petit ARN non codant qu’ils ont baptisé ARN CRISP transactivateur (ARNtracr, Trans-activating CRISPR RNA). Ils ont établi que la séquence de 24 nucléotides de l’ARNtracr est complémentaire de celle des preARNcr, aboutissant à la formation d’un duplex ARNtracr/preARNcr, reconnu par la RNAse III de la bactérie ; en présence de Csn1 (CRISPR-associated Csn1 protein), l’endonucléase introduit des coupures à des sites spécifiques libérant l’ARNcr fonctionnel.
Référence : Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III (2011)
Petits ARN interférents, petits ARN régulateurs et ARN antisens. Depuis leur découverte au début des années 1990, les petits ARN font l’objet d’études intensives. Parmi eux, les petits ARN interférents (pARNi, Small Interfering RNA, siRNA) sont des ARN double brin de petite taille (21 à 24 nucléotides) qui suppriment l’expression d’un gène en activant un mécanisme appelé « interférence ARN » (ARNi). Ce mécanisme de défense permet à la cellule procaryote ou eucaryote de détecter la présence d’un ARN étranger. Il est activé lorsqu’un ARN double brin (ARNdb), par exemple un virus, pénètre dans la cellule. Les ARN interférents sont des régulateurs post-transcriptionnels ; ils guident la dégradation de l’ARNm – correspondant à un gène cible – avec lequel ils possèdent une séquence complémentaire. Les siRNA se lient à un complexe protéique appelé RISC (RNA-Induced Silencing Complex). Le complexe RISC/ARNi scanne les molécules d’ARNm présentes dans la cellule ; en l’absence d’homologie de séquence entre l’ARNi et l’ARNm, celui-ci est traduit en protéine ; dans le cas contraire – homologie de séquence – le complexe RISC se dissocie et libère le brin complémentaire du siRNA, qui se lie à l’ARNm cible. Le composant catalytique du complexe RISC, Argonaute 2, clive l’ARNm cible. Les fragments d’ARNm clivés sont dégradés. Le complexe RISC/ARNi n’est pas détruit et continue à scanner les ARNm. Le complexe RISC/ARNi est devenu un outil d’ingénierie génétique ; connaissant la séquence nucléotidique de l’ARNm d’une protéine particulière, on synthétise un siRNA ayant la capacité de s’hybrider à une séquence de l’ARNm, et on l’introduit dans la cellule.
Anaïs Le Rhun et coll. firent un criblage bio-informatique de la séquence des petits ARN régulateurs (sARN) et ARN antisense (ARNas) chez Streptococcus pyogenes ; les résultats révélèrent que : (i) seuls 20% d’entre eux avaient une fonction connue ; (ii) les endoribonucléases RNase III et RNase Y jouent un rôle essentiel dans la maturation des ARNcr à partir du preARNcr. Pour rappel, ces enzymes sont impliquées dans la dégradation des ARN double brin (RNase III) et des ARNm (RNase Y). La RNase III appartient à une famille de phosphodiestérases très conservées chez les bactéries et les eucaryotes ; elles ont comme cofacteurs des ions métalliques divalents ; sa structure tertiaire révèle la présence d’un « domaine RNase III »qui, en se dimérisant, se lie à l’ARN double brin et hydrolyse les liaisons phosphodiesters sur chaque brin en produisant des fragments aux extrémités 3’-hydroxyle et 5’-phosphodiester caractéristiques avec des surplombs de 2 nucléotides en 3’. L’équipe de Charpentier découvrit une voie nouvelle de maturation des petits ARNcr faisant intervenir l’ARNtracr, la RNase III de Streptococcus pyogenes et l’endonucléase Csn1. C’était la première implication d’un facteur de l’hôte (RNase III) dans l’immunité bactérienne médiée par un ARN.
Références : Le Rhun A, Beer YY, Reimegård J, Chylinski K, Charpentier E RNA sequencing uncovers antisense RNAs and novel small RNAs in Streptococcus pyogenes (2016)
Le Rhun A, Lécrivain AL, Reimegård J, Proux-Wéra E, Broglia L, Della Beffa C, Charpentier E Identification of endoribonuclease specific cleavage positions reveals novel targets of RNase III in Streptococcus pyogenes (2017)
Les endonucléases Cas utilisent des ARN guides. Les Cas (Crispr Associated Protein) forment à une famille de protéines (Cas1, Cas2, Cas3, CasE, Cas6, Csy4, …) dont la plus connue et la plus utilisée en ingénierie génétique est Cas9, l’enzyme clé du système d’immunité adaptative bactérien CRISPR-Cas9 de type II. C’est une endonucléase qui, associée à un petit ARN-guide, introduit des cassures dans l’ADN bicaténaire au niveau d’un site spécifique. Comme il a été dit dans un précédent paragraphe, La découverte de l’ARN CRISP transactivateur (ARNtracr) par Elitza Deltcheva, une étudiante graduée du laboratoire de Charpentier, à Vienne, et ses collègues apporta une pièce essentielle du puzzle ; l’ARNtracr s’apparie à l’ARN CRISPR (ARNcr) (qui contient la séquence virale cible) ; il présente une structure tertiaire en épingle à cheveux ; sa séquence primaire (24 nucléotides) est complémentaire de celle du pré-ARNcr ; l’hybridation de l’ARNtracr et de l’ARNcr forme un complexe ARN bi-hélicoïdal. C’est cet ARN duplex qui guide l’enzyme Cas9 en se liant à la cible d’ADN d’une vingtaine de résidus de longueur, et possédant la séquence Nucléotide – Guanine – Guanine. Cas9 est une protéine de grande taille (1.368 acides aminés) possédant deux domaines nucléasiques distincts : HNH et RuvC. La nucléase HNH coupe le brin complémentaire de l’ADN cible, et la nucléase RuvC-like, le brin non complémentaire.
Références : Chylinski K, Le Rhun A, Charpentier E The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems (2013)
Lécrivain AL, Le Rhun A, Renault TT, Ahmed-Begrich R, Hahnke K, Charpentier E In vivo 3′-to-5′ exoribonuclease targetomes of Streptococcus pyogenes (2018)
D’un colloque à Porto Rico au prix Nobel de chimie. Environ un demi-siècle après l’introduction dans les laboratoires des enzymes de restriction-modification, et seulement trois ans après celle des enzymes de transcription chimériques TALEN, l’ « édition génomique » (genome editing) a subi un formidable coup d’accélérateur lorsque Emmanuelle Charpentier et Jennifer Doudna (University of California, Berkeley) proposèrent d’utiliser, en l’adaptant, le système immunitaire procaryotique CRISPR-Cas9 de type II-A comme outil génétique. A la suite de leur rencontre, en juin 2011, au colloque organisé par l’American Society for Microbiology à San Juan, Porto Rico, Charpentier et Doudna débutèrent une collaboration « courte et intense ». Charpentier et ses collaborateurs soupçonnaient que les petits ARN jouent un rôle dans le mécanisme du système immunitaire bactérien CRISPR de Streptococcus pyogenes ; Jennifer Doudna (Howard Hughes Medical Institute, University of California, Berkeley) était une spécialiste reconnue de l’étude des ARN. Elle consacrait ses efforts à démontrer que « les ARN ne sont pas des spaghettis » mais des entités stables dotées de fonctions catalytiques, au même titre que les protéines. Elle avait publié, avec Thomas Cech, la première structure tridimensionnelle d’un ARN – après celle de l’ARNt -, la structure cristallographique d’un ribozyme de Tetrahymena thermophila.
Référence : Cate JH, Gooding A, Podell E, Zhou K, Golden B, Kundrot C, Cech TR, Doudna JA Crystal structure of a group I ribozyme domain: principles of RNA packing (1996)
Charpentier et Doudna ont cherché à élucider le mécanisme par lequel l’endonucléase Cas9 utilise un ARN pour introduire des coupures ciblées dans un génome ; elles ont démontré que c’est un complexe entre ARNtracr et ARNcr qui guide Cas9 vers un site précis d’un ADN double brin. En conclusion de l’article publié en 2012 par Martin Jinek et coll., Doudna et Charpentier prédisaient : « Our study… highlights the potential to exploit the system for RNA-programmable genome editing.» Cette prédiction s’est pleinement réalisée : elles ont transformé un mécanisme de défense bactérien en « scalpel moléculaire » programmable et polyvalent, capable de cibler avec précision et de couper une séquence d’ADN. Dans un souci de simplification de la technique, Doudna et Charpentier ont fusionné les ARNtracr et ARNcr en une molécule synthétique : l’ARN guide unique (single guide RNA). En changeant la séquence de cet ARN guide, on peut diriger Cas9 pour couper n’importe quelle séquence d’ADN, chez n’importe quel organisme vivant (plantes, animaux, humains). Charpentier et Doudna ont partagé le prix Nobel de chimie 2020.
Note : L’édition génomique – ou modification localisée d’une séquence génomique – est la technologie qui permet de modifier le génome d’une cellule en y insérant, supprimant ou modifiant une séquence d’ADN particulière.
Résumé. Les bactéries et les Archaea possèdent un système de défense immunitaire basé sur une « mémoire », et sur un mécanisme de balayage capable de détecter la présence d’un ADN étranger dans la cellule (un bactériophage, par exemple). La mémoire des infections passées est logée dans une région particulière du génome bactérien appelée locus CRISPR, constitué de séquences répétitives entre lesquelles sont insérés des fragments d’ADN (les « espaceurs ») prélevés sur un virus lors d’infections antérieures des bactéries ou des Archaea. In fine, la cellule neutralise le virus en fragmentant son ADN ; à cette fin, elle mobilise des endonucléases codées par les gènes Cas, regroupés à proximité du locus CRISPR dont il sont séparés par une séquence « leader ». Les protéines Cas sont guidées vers leur cible ADN par un petit ARN guide, dont la séquence reproduit celle d’un espaceur de la bibliothèque génétique des infections passées ; l’espaceur est transcrit en un ARN CRISPR (ARNcr) dont la séquence est complémentaire de celle de la cible ADN, ce qui détermine la spécificité de la coupure double brin par les effecteurs nucléasiques.
Modus Operandi. Idéalement, la séquence cible doit être unique dans le génome, ou à tout le moins représentée à un nombre restreint d’exemplaires ; il en va de la spécificité de la méthode. Les chercheurs sont aidés dans le choix du site répondant à leurs critères par la mise à leur disposition sur le réseau informatique de bibliothèques génomiques où ce type d’information est collectée. Sur le plan expérimental, la mise en œuvre de CRISPR/Cas9 se déroule en deux temps : (i) l’expérimentateur choisit la séquence du guide CRISPR, qui est synthétisé en laboratoire ; la nucléase Cas9 est introduite avec l’ARN guide dans le noyau de la cellule rendue perméable ; (ii) après coupure de l’ADN par la nucléase Cas, le système de réparation de l’ADN de la cellule ressoude la cassure à double brin. C’est à ce stade que l’expérimentateur peut modifier le gène en insérant ou en supprimant des séquences génétiques plus ou moins longues. Le fragment d’ADN est détruit, de même que la nucléase ou les outils moléculaires introduits dans la cellule. Après la réécriture de séquences du génome, on ne peut plus distinguer une mutation naturelle d’une mutation créée par ingénierie génétique. Si la technique permet une sélection précise du site de coupure, la fiabilité du système de réparation de l’ADN est plus aléatoire ; elle varie selon les systèmes biologiques ; elle peut introduire des modifications de séquences imprévues (« effets hors cible »).
Une nouvelle version du système CRISPR-Cas. Les protéines effectrices Cas appartiennent à plus de 45 familles ; elles sont cataloguées en deux types : (i) le type I, monomérique, est représenté par Cas3, une désoxyribonucléase et ATP-hélicase formée d’une seule chaîne polypeptidique ; (ii) le type II, multimérique, est représenté par Cas9, une endo-désoxyribonucléase guidée par un ARN. Le domaine d’application des technologies d’édition génomique basées sur l’emploi de CRISPR s’est encore étendu après la découverte d’une nouvelle famille de nucléases Cas : Cpf1 (CRISPR from Prevotella Francisella1), renommée Cas 12a. Elle a été identifiée chez les bactéries anaérobies Acidaminococcus et Lachnospiraceae par Bernd Zetsche et coll., dans le groupe de Feng Zhang (McGovern Institute for Brain Research, Massachusetts Institute of Technology et Broad Institute, Cambridge), et chez la bactérie pathogène Francisella novicida par Inès Fonfara et coll., dans le groupe d’Emmanuelle Charpentier. Cpf1 est une endoribonucléase et une endonucléase – une dualité fonctionnelle unique parmi les protéines de la famille Cas – mettant en œuvre deux domaines distincts de l’effecteur.
Références : Zetsche B, Gootenberg JS, Abudayyeh O, Slaymaker IM, Makarova KS, Eisenbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System (2015)
Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA (2016)
En 2016, Emmanuelle Charpentier et ses collègues ont présenté une nouvelle version du système CRISPR-Cas mettant en jeu Cpf1. Un certain nombre de particularités de Cpf1 l’ont fait apparaître comme une alternative avantageuse pour remplacer Cas9, l’effecteur le plus utilisé jusque-là : (i) Cpf1 appartient au système CRISPR de Class 2 – comme Cas9 mais, à la différence de Cas9, Cpf1 génère des cassure ADN avec deux brins décalés (les deux domaines nucléasiques HNH et RuvC de Cas9 coupent chacun un brin d’ADN) ; (ii) Cpf1 cible les séquences adjacentes à PAM (Proto-spacer Adjacent Motif) riches en T ; (iii) alors que Cas9 nécessite la présence d’un ARNtrac pour la maturation de l’ARN guide et la coupure de l’ADN, la double activité ARNase et ADNase de Cpf1 lui permet de synthétiser l’ARNcr sans l’aide d’ARNtracr ; (iv) en n’utilisant qu’un seul ARNcr court (/- 42 nucléotides), le système CRISP-Cpf1 permet une édition plus précise et le ciblage simultané de plusieurs gènes (multiplexage). Charpentier a qualifié la nouvelle version du système CRISPR-Cas basée sur l’emploi de Cpf1, de « most minimalistic so far described ».
Le domaine d’application des outils d’édition CRISPR-Cas9 et CRISPR-Cpf1 est très vaste ; leur succès est attesté par le fait qu’on recensait en 2025, plus de quinze mille publications scientifiques faisant mention de leur emploi. Pour ne citer que quelques-unes des applications possibles : régulation transcriptionnelle, modulation épigénétique (par méthylation de bases de l’ADN, modification des histones), édition de bases, criblage à grande échelle, détection d’agents pathogènes (SARS-CoV-2), génétique fonctionnelle (découvrir la fonction d’un gène) génotypage multiplexe (l’expression simultanée de plusieurs ARN guides et enzymes Cas). L’ADN peut être modifié en plusieurs endroits en même temps, en série, et des familles entières de gènes peuvent être désactivées dans un organisme.
Références : Cong L, Ran A, Cox D, Lin S, Baretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zang F Multiplex Genome Engineering Using CRISPR/Cas Systems (2013)
Jiang W, Bikard D, Cox D, Zang F, Marraffini LA RNA-guided editing of bacterial genomes using CRISPR-Cas systems (2013)
Safari F, Zare K, Negahdaripour M, Barekati-Mowahed M, Ghasemi Y CRISPR Cpf1 proteins: structure, function and implications for genome editing (2019)
En 2014, la physicienne Eva Nogales (Lawrence Berkeley National Laboratory), spécialiste de la cryomicroscopie électronique, collabora avec Doudnapour établir la structure atomique tridimensionnelle de Cas9. En se fixant sur la séquence cible d’ADN, l’ARN-guide induit un changement conformationnel de cette endonucléase, avec formation d’un tunnel autour de l’acide nucléique.
Références : Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E Doudna JA Structures of Cas9 endonucleases reveal RNA-mediated conformational activation (2014)
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA A three-dimensional view of the molecular machinery of RNA interference (2014)
Note : Après six ans de stages post-doctoraux aux Etats-Unis, Emmanuelle Charpentier s’est heurtée à la difficulté pour un jeune chercheur de trouver une structure d’accueil et un financement pour créer sa propre équipe de recherche en France. Ce ne fut pas le cas en Autriche où elle trouva environnement scientifique international et financement (Institut für Mikrobiologie und Genetik, Department of Microbiology and Immunobiology, Universität Wien, Max F. Perutz Laboratories, Wien). Puis, la Suède lui a offert un financement substantiel et une liberté totale dans le choix de ses thèmes de recherches (The Laboratory for Molecular Infection Medicine Sweden, Umeå Centre for Microbial Research, Department of Molecular Biology, Umeå University). Enfin, en Allemagne, la prestigieuse Max Planck-Gesellschaft lui a permis de bénéficier de ses conséquents budgets de recherche et de son absence de bureaucratie (Helmholtz Centre for Infection Research, Braunschweig et Hannover Medical School ; Max Planck Institute for Infection Biology, Berlin). En 2018, la Max Planck-Gesellschaft a créé un institut indépendant (Max-Planck-Forschungsstelle für die Wissenschaft der Pathogene) sur le campus Charité Mitte Berlin, dont Charpentier assure la direction.
TALEN
Les protéines TALE reconnaissent les bases nucléotidiques de l’ADN. Les « pseudo-activateurs de transcription, Transcription activator-like effectors (TALE) » sont des protéines dérivant de protéines de bactéries phytopathogènes du genre Xanthomonas ; ils augmentent la transcription des gènes en se liant à l’ADN ; ils imitent ainsi l’action des activateurs de transcription de la cellule mais ils s’en différencient par leur incapacité à déclencher efficacement la transcription ou à la mener à son terme. Le mécanisme par lequel les pseudo-activateurs de transcription, après avoir pénétré dans une cellule végétale, se fixent sur l’ADN de l’hôte a été décrit par Jens Boch et coll. (Department of Genetics, Institute of Biology, Martin-Luther-University Halle-Wittenberg) en 2009. Les pseudo-activateurs de transcription ont une structure modulaire constituée de séquences répétitives de 33 à 34 acides aminés. Chacune de ces unités répétitives reconnaît une paire de bases l’ADN (A, T, C ou G) selon un code de reconnaissance dicté par leur séquence en acides aminés. Matthew J. Moscou et Adam J. Bogdanove (Department of Plant Pathology and Bioinformatics and Computational Biology Program, Iowa State University) ont montré le rôle prédominant du duo d’acides aminés situés en positions 12 et 13 de la séquence d’une unité répétitive. Le code de reconnaissance une fois déchiffré ouvre la possibilité de fabriquer des protéines TAL reconnaissant une séquence génomique déterminée.
Références : Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U Breaking the code of DNA binding specificity of TAL-type III effectors (2009)
Moscou MJ Bogdanove AJ A simple cipher governs DNA recognition by TAL effectors (2009)
Des nucléases chimériques. Un groupe de chercheurs sous l’égide d’Edward J Rebar (Sangamo BioSciences, Inc., Richmond, California) a tiré partie du code de reconnaissance des activateurs de transcription entrepris pour concevoir des nucléases spécifiques, actives dans les cellules végétales et animales. Pour construire les TALEN (Transcription Activator-Like Effector Nuclease), ils ont associé un domaine TALE à l’endonucléase FokI. A la différence de la majorité des endonucléases, FokI est composée de deux domaines fonctionnels : un domaine de reconnaissance de l’ADN et un domaine catalytique. Ils ont remplacé le domaine de reconnaissance de la nucléase par un domaine TALE assemblé en laboratoire. FokI est un enzyme monomérique mais c’est sous forme de dimère qu’il coupe les deux brins d’ADN. Pour cibler une séquence spécifique dans un génome, il est nécessaire de créer une sonde réunissant plusieurs protéines TAL – orientées tête-bêche. La longueur du segment d’acides-aminés séparant le domaine TALE de FokI, et le nombre de nucléotides cibles sur le génome, doivent être définis avec précision. En pratique, on utilise deux sondes qui se fixent face à face sur les deux brins de l’ADN, permettant aux enzymes FokI de se rejoindre et de sectionner la double hélice. On a encore accru la spécificité et l’efficacité de FokI en fabriquant, par ingénierie génétique, des versions améliorées par mutations. La cassure introduite par TALEN est réparée par les enzymes de la cellule, ce qui n’exclut pas le risque de mutations potentielles. Lors de la réparation, les chercheurs peuvent introduire, modifier ou supprimer un gène.
L’emploi de la technologie TALEN a été éclipsée par l’arrivée de la technologie CRISPR-Cas9, plus facile et moins coûteuse. Il existe cependant deux domaines où elle garde son intérêt pour sa finesse et sa flexibilité : la génétique classique emprunte la voie allant des gènes au phénotype (protéines, ARN). En génétique inverse, on cherche à élucider la fonction d’un ou de plusieurs gènes en partant de variations de phénotypes dans les cellules ou les organismes. Pour cette approche, les TALEN permettent de modifier un gène spécifique avec plus de précision que les autres méthodes d’édition génomique. Dans la technologie TALEN, ce sont deux longues protéines qui se fixent sur l’ADN réduisant ainsi le risque de modifications « hors-cible ». D’autre part, l’emploi de la technologie TALEN est plus étendu parce qu’elle fait l’impasse sur la présence d’une séquence PAM à proximité de la cible, comme dans le système CRISPR ; les TALEN, n’ayant pas cette contrainte, peuvent cibler n’importe quelle séquence d’ADN. L’autre domaine où la technologie TALEN garde tout son intérêt est la thérapie génique humaine ; la grande spécificité de cette technologie permet à l’expérimentateur de modifier un gène spécifique avec plus de précision que les autres méthodes d’édition génomique. TALEN a été utilisé pour modifier des séquences d’ADN de nombreux organismes modèles, des bactéries aux mammifères.
Références : Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ A TALE nuclease architecture for efficient genome editing (2011)
Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P Efficient construction of sequence-specific TAL (2011)
Le domaine d’application des technologies d’édition génomique basées sur l’emploi de CRISPR s’est encore étendu après la découverte d’une nouvelle famille de nucléases Cas : Cpf1 (renommée Cas 12a). Elle a été identifiée chez les bactéries anaérobies Acidaminococcus et Lachnospiraceae – par Bernd Zetsche et coll., dans le groupe de Feng Zhang (McGovern Institute for Brain Research, Massachusetts Institute of Technology et Broad Institute, Cambridge) – et chez la bactérie pathogène Francisella novicida – par Inès Fonfara et coll., dans le groupe d’Emmanuelle Charpentier. Le sigle Cpf1 vient de CRISPR from Prevotella Francisella1.
Références : Zetsche B, Gootenberg JS, Abudayyeh O, Slaymaker IM, Makarova KS, Esslebichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System (2015)
Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA (2016)
On connaissait déjà de nombreuses protéines effectrices Cas (CRISPR-associated proteins) appartenant à plus de 45 familles, et cataloguées en type I, monomérique, représenté par Cas3, une désoxyribonucléase et ATP-hélicase formée d’une seule chaîne polypeptidique, et le type II, multimérique, représenté par Cas9, une endo-désoxyribonucléase guidée par un ARN. En 2016, Emmanuelle Charpentier et ses collègues de la Max Planck Unit for the Science of Pathogens, Berlin, du Laboratory for Molecular Infection Medicine Sweden, Umeå University, et du Helmholtz Centre for Infection Research, Braunschweig, ont présenté une nouvelle version du système CRISPR-Cas mettant en jeu Cpf1, une endoribonucléase et endonucléase, une dualité fonctionnelle unique parmi les protéines de la famille Cas. Les deux réactions mettent en œuvre des domaines distinct de l’effecteur. Un certain nombre de particularités de Cpf1 l’ont fait apparaître comme une alternative avantageuse pour remplacer Cas9, l’effecteur le plus utilisé jusque-là : comme Cas9, Cpf1 appartient aux systèmes CRISPR de Class 2 ; à la différence de Cas9, Cpf1 génère des cassure ADN avec deux brins décalés. Comme Cas9, cette endonucléase reconnaît un motif PAM (Proto-spacer Adjacent Motif) riche en T, mais alors que Cas9 nécessite la présence d’un ARNtrac pour la maturation de l’ARN guide et la coupure de l’ADN, la double activité ARNase et ADNase de Cpf1 lui permet de synthétiser l’ARNcr sans l’aide d’ARNtracr. Je rappelle que la reconnaissance de la séquence cible sur l’ADN implique (i) la complémentarité des bases entre cette séquence et celle de l’ARNcr ; (ii) la présence d’une courte séquence PAM de 2 à 6 pb (5′-Y-T-N-3′ dans le cas de Cpf1), située sur le brin ADN qui ne contient pas la séquence cible (8 nucléotides). Les séquences de l’ARNcr et de PAM définissent avec précision le site de coupure. L’ARNtrac, apparié à une partie de l’ARNcr forme une épingle à cheveu, qui sert à la nucléase de repère d’ancrage sur l’ADN. La reconnaissance de la séquence PAM provoque une fusion localisée de l’ADN double brin, permettant à l’ARNcr d’accéder au brin d’ADN auquel il s’apparie. Comme il a été dit précédemment, les effecteurs Cas sont formés de deux domaines nucléasiques, HNH et RuvC, qui coupent chacun un brin d’ADN ; dans le cas de Cpf1 la coupure génère deux extrémités décalées de 5 à 8 nucléotides. Emmanuelle Charpentier a qualifié la nouvelle version du système CRISPR-Cas basée sur l’emploi de Cpf1, de « most minimalistic so far described ».
Références : Jiang W, Bikard D, Cox D, Zang F, Marraffini LA RNA-guided editing of bacterial genomes using CRISPR-Cas systems (2013)
Safari F, Zare K, Negahdaripour M, Barekati-Mowahed M, Ghasemi Y CRISPR Cpf1 proteins: structure, function and implications for genome editing (2019)
Le locus CRISPR-Cpf1 est constitué de 9 espaceurs séparés par des séquences répétitives longues de 36 nucléotides. Dans les familles de protéines des bactéries anaérobies Acidaminococcus et Lachnospiraceae, on a identifié deux orthologues Cpf1 utilisables pour l’édition génomique de cellules humaines. Le domaine d’application des outils d’édition CRISPR-Cas9 et Cpf1est très vaste ; leur succès est attesté par le fait que, depuis leur introduction, on recense, aujourd’hui (2025), plus de quinze mille publications scientifiques faisant mention de son emploi. Pour ne citer que quelques-unes des applications possibles : régulation transcriptionnelle, modulation épigénétique (par méthylation de bases de l’ADN, modification des histones), édition de bases, criblage à grande échelle, détection d’agents pathogènes (SARS-CoV-2), génétique fonctionnelle (découvrir la fonction d’un gène) génotypage multiplexe (l’expression simultanée de plusieurs ARN guides et enzymes Cas). L’ADN peut être modifié en plusieurs endroits en même temps en série et des familles entières de gènes peuvent être désactivées dans un organisme.
Références : Cong L, Ran A, Cox D, Lin S, Baretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zang F Multiplex Genome Engineering Using CRISPR/Cas Systems (2013)
Jiang W, Bikard D, Cox D, Zang F, Marraffini LA RNA-guided editing of bacterial genomes using CRISPR-Cas systems (2013)
Prime editing
Il était dit que dans le domaine de l’édition génomique nous irions de révolution en progrès spectaculaires. En 2019, le groupe de David R. Liu (Merkin Institute of Transformative Technologies in Healthcare, Broad Institute of Harvard and MIT, Cambridge) proposa un nouvel outil – le « Prime editing », encore appelé « Base editing » – offrant sur CRISPR-Cas le double avantage d’être plus précis dans les modifications apportées à l’ADN et de mieux les contrôler ; les mécanismes de réparation de l’ADN intervenant après la coupure introduisent un risque d’erreur. Tant que le problème des effets hors-cible (off-target), introduisant des modifications collatérales dans le génome, n’est pas résolu, la possibilité d’utiliser CRISPR-Cas en clinique est limitée. Le risque d’effets « hors cible » n’existe pas avec le Prime editing puisqu’il n’y a ni coupure des deux brins de l’ADN, ni nécessité d’utiliser une matrice ADN. A la stratégie « couper puis coller » de CRISPR-Cas – supprimer une séquence d’ADN et la remplacer par une autre – on substitue une stratégie « chercher puis remplacer » : modifier base par base le code génétique.
Référence : Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR Search-and-replace genome editing without double-stranded breaks or donor DNA (2019)
La mise en œuvre du Prime editing commence par la fabrication d’un outil moléculaire constitué de la Prime editor protein et du Prime editing guide RNA. Ce dernier – acronyme l’ARNpeg – a une double fonction : (i) sélectionner la séquence cible ADN (Primer binding site) ; celle-ci est spécifiée par un ARN dont la séquence est complémentaire ; (ii) spécifier la modification à introduire ; c’est le rôle dévolu à une matrice (template) associée. Cette longue molécule renferme successivement, de l’extrémité 5’ à l’extrémité 3’ : une séquence guide ancrée dans la nickase par une structure en épingles à cheveux, un segment d’ARN ancré dans la transcriptase réverse par une seconde structure en épingles à cheveux ; l’extrémité 3’ de l’ARNpeg avec la matrice et la séquence guide (Primer binding site) émerge du complexe. Pour la « Prime editor protein » on utilise un complexe associant une nucléase Cas9 dont on a diminué la capacité catalytique de façon à ce qu’elle n’entaille qu’un seul brin d’ADN (nickase), en créant une extrémité 3’ libre, et une transcriptase réverse, produite par ingénierie, qui utilise la partie matrice de l’ARNpeg pour synthétiser un très court fragment d’ADN monobrin de séquence définie par l’expérimentateur. Cette réparation se déroule sur le Primer binding site.
Le Prime editing est un outil d’édition génomique versatile pour modifier de façon très précise le génome d’organismes vivants. Son apparition a été largement diffusée dans la presse non scientifique à cause des possibilités qu’il offre dans le domaine des maladies génétiques chez l’homme, en rendant possible la correction des mutations génétiques ex-vivo ou in vivo.
Référence : Liu P, Liang SQ, Zheng C, Mintzer E, Zhao YG, Ponnienselvan K, Mir A, Sontheimer EJ, Gao G, Flotte TR, Wolfe SA Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice (2021)
Les transposons sont des éléments génétiques – des séquences d’ADN endogènes – capables de se déplacer au sein d’un génome. Ils sont largement répandus chez les animaux, les végétaux et les bactéries ; ils peuvent aussi être introduits dans les cellules à l’aide d’un vecteur pour provoquer des mutations. Les rétrotransposons se différencient des transposons par l’intermédiaire utilisé: un ARN au lieu d’ADN. Cet ARN est transcrit en ADN par la réverse transcriptase. Pour se déplacer et s’insérer dans le génome à l’aide d’une transposase, les éléments génétiques mobiles ont recours aux mécanismes du « couper puis coller » (transposons) ou du « copier puis coller » (rétrotransposons). Il existe donc dans la nature des mécanismes permettant d’insérer de larges fragments d’ADN dans un génome.
Les systèmes de Prime editing souffrent d’une limitation: la longueur des fragments d’ADN qu’elles peuvent insérer dans un génome n’excède pas 66 pb avec une efficacité de 20%. C’est un handicap lorsqu’on veut traiter des maladies génétiques en remplaçant un gène défectueux. Pour palier à cet inconvénient, Jin Wang (Medical Research Institute, Zhongnan Hospital of Wuhan University) et un groupe de collaborateurs appartenant à 9 institutions ont inventé le GRAND editing, permettant l’insertion directe et précise dans un génome de fragments d’ADN allant jusqu’à 1 kb. L’originalité de cette méthode tient au fait qu’au lieu d’utiliser un donneur ADN, on mobilise deux ARNpeg dont les matrices (templates) ne sont pas homologues à l’ADN cible (Primer binding site) mais partiellement complémentaires l’une de l’autre, si bien qu’elles peuvent s’apparier en une structure bi-hélicoïdale, qui sera transcrit par la réverse transcriptase en ADN double brin stable. Celui-ci représente le fragment d’ADN dont l’intégration ciblée dans le génome remplacera le fragment de gène défectueux. Sa longueur peut atteindre 150 pb (efficacité 63%), 250 pb (efficacité 28%) et jusqu’à 1000 pb (efficacité 1%). On perçoit immédiatement les avantages d’une telle approche expérimentale en thérapeutique.
Comme dans la méthode classique d’édition génomique, les deux ARNpeg dirigent la nickase vers les séquences cibles d’ADN double brin ; la nucléase pratique deux entailles sur le brin opposé ; les ARNpeg servent alors de matrices pour la réverse transcriptase qui synthétise deux ADN monocaténaires qui s’apparient du fait de la complémentarité partielle de leurs séquences. Ce nouveau fragment entre en compétition avec le gène défectueux ; si il est intégré dans le génome, il est pris en charge par la machinerie cellulaire de réparation de l’ADN.
Références : Wang J, He Z, Wang G, Zhang R, Duan J, Gao P, Lei X, Qiu H, Zhang C, Zhang Y, Yin H Efficient targeted insertion of large DNA fragments without DNA donors (2022) Zheng C, Liu B, Dong X Gaston N, Sontheimer EJ, Xue W Template-jumping prime editing enables large insertion and exon rewriting in vivo (2023)