Mitochondries
Sur la planète Terre, environ 90% de l’oxygène consommé par les êtres vivants est utilisé dans des organites subcellulaires appelés « mitochondries ». On en trouve les premières représentations dans un ouvrage publié en 1841 par Friedrich G. J. Henle, un élève de Johannes Peter Müller, titulaire de la chaire de physiologie et d’anatomie de la Friedrich-Wilhelm Universität, à Berlin. Les cellules musculaires ont d’importants besoins en énergie rapidement mobilisable ; les mitochondries sont les seuls organites capables de remplir cette fonction, ce qui explique pourquoi les tissus musculaires en contiennent de grandes quantités. Élève de Lorenz Oken, le pape de la Naturphilosophie, et de Johannes Peter Müller, Rudolph A. von Kölliker fut professeur de physiologie et d’anatomie comparée à l’Université de Wurtzbourg. Microscopiste expérimenté, il observa, en 1857, dans le sarcoplasme des muscles striés, des granules interstitiels individualisés qu’il appela körperchen (corpuscules) sui generis ». En 1890, Richard Altmann, professeur d’anatomie à l’Université de Leipzig, mit au point une technique de fixation et de coloration des tissus à l’aniline et fuschine ; il détecta la présence de « bioblastes » dans le cytoplasme des cellules. Walther Flemming, professeur d’anatomie à l’Université de Kiel, observa des « filae » dans une variété de tissus. L’utilisation d’un nouveau colorant, le cristal violet, permit à Carl Benda de détecter la présence de ces organites dans des tissus où ils sont peu abondants. Les microscopistes utilisèrent la coloration dite « vitale » au Vert Janus de Leonor Michaelis, plus connu pour avoir, avec Maud L. Menten, proposé en 1913 l’équation décrivant la cinétique des réactions enzymatiques. Au contact du colorant, les organites prennent une teinte bleu foncé ce qui révéla la présence de réactions d’oxydation. Les mitochondries furent d’abord appelées « granules de Kölliker », filae, bioblastes. En 1890, Retzius introduisit l’appellation « sarcosome » pour désigner les organites présents dans les muscles. En 1898, Carl Benda créa le terme « mitochondrie » par référence à la forme allongée de l’organite (mitos = fil). En 1909, Claudius Regaud (Faculté de médecine, Lyon) mit au point une technique de coloration des mitochondries et montra que « sarcosome » et « mitochondrie » désignent la même entité subcellulaire. Le terme « chondriosome », pour désigner les mitochondries, fut employé jusqu’au milieu du XXe siècle.
Références : Henle FGJ Allgemeine Anatomie: Lehre von den Mischungs-und Formbestandtheilen des Menschlichen Körpers (1841)
Altmann R Die Elementarorganismen und ihre Beziehungen zu den Zellen (1890)
Flemming W Zellsubstanz, Kern und Zellteilung (1882)
Benda C Ueber die Spermatogenese der Vertebraten und höherer Evertebraten, II Theil: Die Histiogenese der Spermien (1898)
Plusieurs hypothèses furent avancées concernant la nature et la fonction de ces organismes cytoplasmiques. Pour certains, il s’agissait de bactéries. Pour Richard Altmann, les « bioblastes » étaient des particules vivantes élémentaires dotées d’une autonomie métabolique et génétique, et capables de se reproduire ; c’était prémonitoire ! Le botaniste Andreas F.W. Schimper fit remarquer que les chloroplastes des cellules végétales présentent de nombreuses similarités avec les cyanobactéries. Les observations de Schimper ont inspiré le botaniste Konstantin S. Mereschkowski, professeur de microbiologie (Université fédérale de Kazan). En 1905, une vingtaine d’années après les travaux de Schimper, Mereschkowski inventa la « symbiogenèse » comme conclusion à ses travaux sur les lichens : des êtres primitifs (champignons, algues, lichens) peuvent s’associer pour former des êtres plus complexes. Il émit l’hypothèse que la capture de cyanobactéries par des protozoaires a conduit à l’apparition des chloroplastes des cellules végétales. Si l’article en allemand, qu’il publia en 1909, recueillit peu d’échos parmi les scientifiques, Ivan E. Wallin, professeur d’Anatomie (Medical School, University of Colorado) en eut connaissance. Il appliqua les déductions de Mereschkowski au cas des mitochondries. En 1922, et dans les années suivantes, il publia une série d’articles dans l’American Journal of Anatomy démontrant, sur des bases expérimentales, que les mitochondries proviennent de bactéries endocytées par la cellule primitive. Cette affirmation fut étayée lorsque l’examen au microscope électronique révéla la présence de ribosomes dans la matrice mitochondriale, donc d’un appareil de traduction. En 1980, la découverte de l’ADN mitochondrial (ADNmt) mit un point final à la controverse sur l’origine des mitochondries.
La théorie endosymbiotique est l’œuvre de la bactériologiste Lynn Margulis (Boston University). Cette théorie est une remise en cause radicale de la « théorie synthétique » chère aux néo-darwinistes du début du XXe siècle : les mutations génétiques et la sélection naturelle sont les seuls facteurs à l’origine de l’apparition de nouvelles espèces. Inutile de préciser que Margulis éprouva les plus grandes difficultés à publier le résultat de ses travaux ; bien que n’étant à l’époque qu’une enseignante en début de carrière, elle eut le mérite de tenir tête au désaveu de l’establishment universitaire et scientifique. Après une quinzaine de refus ! Son article fut accepté par le comité éditorial du Journal of Theoretical Biology ; il parut en 1966. Quinze plus tard, Margulis résuma le résultat de ses travaux dans un ouvrage qui parut en 1981 : les variations héréditaires, qui sont le moteur de l’évolution, ne proviennent pas seulement de mutations aléatoires ; il y a aussi une évolution par associations d’organismes primitifs (une α-protéobactérie aérobie avec une cellule primitive dans le cas des mitochondries). La cellule eucaryote est la composante d’une série d’associations symbiotiques avec des procaryotes capturés par la cellule primitive.
Les mitochondries se divisent en même temps que la cellule. Le génome mitochondrial est un génome compact, sans introns, dont l’ADN (comme celui des chloroplastes) présente de grandes similitudes avec l’ADN bactérien : il est souvent bicaténaire et circulaire. Les hépatocytes renferment environ 4.000 mitochondries contenant 2 à 4 molécules d’ADNmt, par mitochondrie. La séquence de l’ADNmt chez la souris, l’homme et le bœuf fut établie par le groupe du MRC Laboratory of Molecular Biology, Cambridge (16.569 paires de bases pour l’ADNmt humain). Des erreurs de séquence furent corrigées en 1999. Les 37 gènes de l’ADNmt codent principalement pour des ARN (22 ARNt 2 ARNr) et 13 protéines.
Références : Altmann R Die Elementarorganismen und ihre Beziehungen zu den Zellen (1890)
Schimper AFW Ueber die Entwickelung der Chlorophyllkörner und Farbkörper Ueber Chlorophyllkörner Stärkebildner und Farbkörper (1883)
Mereschkowski K Über Natur und Ursprung der Chromatophoren im Pflanzenreiche (1905)
Mereschkowski K Theorie der zwei Plasmaarten als Grundlage der Symbiogenesis, einer neuen Lehre von der Entstehung der Organismen (1910)
Wallin IE On the Nature of Mitochondria (1922)
Wallin IE Symbionticism and the Origin of Species (1927)
Margulis L The Origin of Mitosing Eukaryotic Cells (1966)
Margulis L Symbiosis in Cell Evolution (1981)
Margulis L Archaeal-eubacterial mergers in the origin of Eukarya: phylogenetic classification of life (1996)
Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJH, Staden R, Young IG Sequence and organization of the human mitochondrial genome (1981)
Des « granules respiratoires »
En 1789, dans son laboratoire de l’Arsenal, à Paris, Antoine Laurent de Lavoisier et son disciple Armand Séguin montrèrent que l’oxygène est indispensable à la combustion et, chez les animaux, à la respiration. Chez ces derniers, la consommation d’oxygène s’accompagne d’une production de gaz carbonique. Malheureusement pour Lavoisier, il cumulait avec sa fonction de régisseur des poudres et salpêtres la charge de fermier général, c’est-à-dire de collecteur d’impôts ; il fut arrêté pendant la Terreur et traduit devant le tribunal révolutionnaire qui, décrétant que « la République n’a pas besoin de savants », envoya à la guillotine le plus grand chimiste de son temps.
A quoi sert l’oxygène inspiré par nos poumons ? Où va-t-il exactement ? En 1912, l’histologiste Benjamin F. Kingsbury émit l’hypothèse que l’oxygène fourni par la respiration est consommé dans les mitochondries. Ainsi naquit la notion de « respiration cellulaire », une appellation qui désigne l’ensemble des processus métaboliques qui permettent à un être vivant de tirer des nutriments qu’il ingurgite l’énergie nécessaire à ses besoins vitaux. La respiration cellulaire fut le grand sujet d’études des biochimistes du début du XXe siècle. En 1910, alors qu’il était étudiant dans le laboratoire d’Hermann E. Fischer (prix Nobel de chimie en 1902), à l’Institut für Chemie, Université de Berlin, Otto Warburg découvrit que des granules séparés par centrifugation d’un broyat tissulaire catalysent l’oxydation d’acides organiques en présence de cytochrome c. Il avait mis au point un appareil manométrique pour mesurer les échanges gazeux – par exemple, la consommation d’oxygène par des coupes de tissus, en présence de substrats organiques. La préparation de bonnes coupes de tissus est difficile ; compte tenu du coefficient de diffusion de l’oxygène et de la vitesse à laquelle il est consommé par les tissus, l’épaisseur des tranches ne devait pas dépasser une dizaine de couches de cellules. L’utilisation de broyats ou de fractions subcellulaires permettait d’éviter cet inconvénient.
Des tentatives pour isoler les granules dotés d’activité respiratoire (des mitochondries selon l’hypothèse de Kingsbury) furent entreprises en 1933 par Robert R. Bensley et Normand L. Hoerr (The University of Chicago). Bensley était connu pour ses travaux sur le pancréas endocrine : il avait démontré que les îlots de Langerhans constituent un tissu spécialisé, et mis au point une technique de coloration pour distinguer les cellules α des cellules β. Bensley développa une laborieuse technique de purification des mitochondries ; les tissus étaient congelés immédiatement après prélèvement et broyés ; l’homogénat était centrifugé à travers un gradient de densité de solvants organiques (pour limiter la perte par diffusion de molécules hydrosolubles). Les organites ainsi obtenus étaient altérés par l’action des solvants sur les phospholipides membranaires. Vers le milieu des années 1940, Albert Claude et son technicien Rollin Hotchkiss (Rockefeller Institute for Medical Research) utilisèrent une solution de chlorure de sodium pour préparer l’homogénat tissulaire ; ils isolèrent par centrifugation différentielle une fraction de « Gros granules ». A l’examen microscopique, cette fraction contenait des mitochondries altérées. Les solutions salines provoquent l’agrégation des particules subcellulaires. En 1946, Walter Schneider et George Hogeboom remplacèrent la solution de chlorure de sodium par de l’eau distillée : le résultat ne fut pas meilleur ; les organites de forme sphérique ainsi isolés n’étaient pas colorés par le Vert Janus (le colorant vital des mitochondries) et n’avaient pas leur morphologie allongée caractéristique. A la suggestion de George Palade, Hogeboom et Schneider remplacèrent l’eau par une solution de saccharose et le broyage des tissus au mortier et au pilon par l’emploi de l’homogénéiseur en verre mis au point en 1936 par Van Rensselaer Potter et Conrad Arnold Elvejehm (Department of Agricultural Chemistry, University of Wisconsin, Madison). Les organites isolés avaient une activité respiratoire et la morphologie des mitochondries observées au microscope.
Références : Bensley RR, Hoerr NL Studies on cell structure by the freezing-drying method. VI. The preparation and properties of mitochondria(1934)
Hogeboom GH, Schneider WC, Pallade GE The Isolation of Morphologically Intact Mitochondria from Rat Liver (1947)
Il fallut attendre l’arrivée du microscope électronique pour découvrir la structure fine des mitochondries. Une compétition s’établit entre le groupe du Rockefeller Institute à New York et celui du Karolinska Institutet, à Stockholm. Après le départ d’Albert Claude du « Rockefeller Institute », les group leaders étaient Keith Porter et George Palade. En microscopie électronique, le laboratoire de New York avait fait œuvre de pionnier. Claude et Porter avaient publié la première micrographie de cellule (un fibroblaste étalé sur une grille) et Claude et Ernest Fullam, le premier cliché d’organites, en 1945. A Stockholm, le physicien Karl Manne G. Siegbahn, prix Nobel de physique en 1924, directeur de l’Institut Nobel de Physique, avait commencé à s’interresser à la microscopie électronique avant la Seconde Guerre mondiale. Siegbahn suivait de près les efforts de ses voisins allemands du groupe d’Ernst Ruska pour mettre au point un microscope électronique. En 1938, Siegbahn décida que la Suède devait entrer dans la compétition et chargea son assistant, Fritiof S. Sjöstrand, d’étudier les applications en biologie de la microscopie électronique. Sjöstrand avait acquis la pratique de la microscopie dans le département de pharmacologie du Karolinska Institutet. Les groupes de New York et de Stockholm se trouvèrent vite en concurrence. Leur différent porta principalement sur les points suivants : la mitochondrie est-elle entourée d’une simple ou d’une double membrane ? Quel est le rôle des cristae ? Sont-elles en continuité avec la membrane interne ?
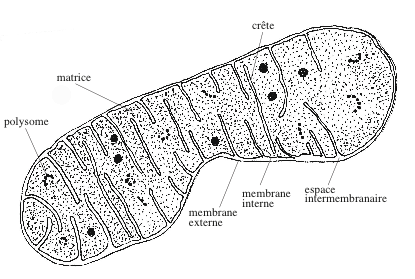 |
| Mitochondrie. C’est un organite allongé, entouré d’une enveloppe constituée d’une double membrane. Entre la membrane externe et la membrane interne, se trouve un espace intermembranaire de 8 à 10 nanomètres d’épaisseur. La membrane interne forme des invaginations, les crêtes mitochondriales ou cristae. La membrane externe est perméable aux molécules de masse inférieure à 500, aux ions et aux protons. La membrane interne est perméable aux petites molécules (O2, H2O, CO2, NO, NH3) et imperméable aux protons. Elle renferme des transporteurs, une chaîne de transfert d’électrons et l’ATP synthase. La matrice a une apparence finement granuleuse. Elle héberge de l’ADN, un appareil de transcription et de traduction, des polysomes, des concrétions minérales, les enzymes du cycle de Krebs, de la β-oxydation des acides gras et de l’oxydation des acides aminés. |
En 1943, Sjöstrand publia dans la revue Nature les premières micrographies montrant la double membrane. Celles qu’il montra en 1952 à la réunion de l’Electron Microscopy Society of America, étaient plus convaincantes ; on y voyait les cristae, des cloisons transversales qui, selon Sjöstrand, divisaient la matrice mitochondriale en compartiments. C’était un point de désaccord avec Palade pour qui les cristae, percées de pores, n’interrompent pas la continuité de la matrice. A la Third International Conference on Electron Microscopy qui se tint à Londres en 1954, Sjöstrand persista dans l’erreur en soutenant que les pores observés par Palade dans les crêtes étaient des artefacts de conservation des échantillons.
Référence: Sjöstrand F. Electron-microscopic examination of tissues (1943)
Atmungsferment ou déshydrogènases ?
Comme je l’ai dit plus haut, la respiration cellulaire – c’est-à-dire la consommation d’O2 par les tissus – était, au début du XXe siècle, un thème de recherches à la mode. La technique expérimentale consistait à mettre le tissu – un muscle par exemple – en contact avec un substrat et à mesurer la consommation d’O2 (ou la production de CO2) à l’aide de manomètres plus ou moins élaborés. La respiration tissulaire a d’abord été étudié sur des organes entiers, le plus souvent des muscles, puis sur des tranches minces de tissus (0,2 à 0,5 mm d’épaisseur), dans des broyats cellulaires, et dans le système acellulaire mis au point par Batelli et Stern. Elle attira des biochimistes de renom ; en 1910, Otto Warburg avait découvert l’Atmungsferment associé à des granules subcellulaires. Heinrich O. Wieland, professeur de chimie à l’université de Munich, prix Nobel de chimie en 1927, étudiait la réduction de substances organiques in vitro, en présence de catalyseurs chimiques, ou en milieu cellulaire. En 1913, il découvrit que la bactérie Acetobacter convertit l’éthanol en acétate en présence d’un accepteur d’électrons (bleu de méthylène ou une quinone) :
CH3-CH2OH ➔ CH3-COOH
La réaction se déroulant en l’absence d’oxygène, il en déduisit qu’elle était catalysée par une déshydrogénase. En effet, l’éthanol cède deux atomes d’hydrogène qui ont la capacité de réduire de l’oxygène pour former une molécule d’eau :
O2 + 2 H2 ➔ 2 H2O
L’éthanol est plus riche en hydrogène que l’acétate. Dans la réaction (non équilibrée) écrite plus haut, l’éthanol est la forme réduite et l’acétate, la forme oxydée. Pour dire les choses autrement, la réaction redox (réduction/oxydation) :
H2 ↔ 2H+ + 2e–
indique que le transfert d’une molécule d’hydrogène équivaut au transfert de deux électrons. L’éthanol, plus riche en électrons que l’acétate, sert de « donneur » et l’acétate, de « receveur ». Les êtres vivants hétérotrophes tirent leur énergie des électrons fournis par les nutriments.
Dans les années 1900 à 1910, Torsten L. Thunberg (Lunds Universitet) étudia la capacité de tranches de tissus, placées dans un milieu de survie, à oxyder des acides organiques en présence d’un accepteur d’électrons. La consommation d’oxygène de broyats cellulaires était mesurée par la vitesse de décoloration du bleu de méthylène dans un « tube de Thunberg ». Il sélectionna une soixantaine de sels d’acides carboxyliques : lactate, pyruvate (un groupe carboxylique), succinate, malate, fumarate, glutamate (deux groupes), citrate (trois groupes). La vitesse de consommation de l’O2 dépendait de la nature du substrat. Apportant de l’eau au moulin d’Heinrich Wieland, Thunberg conclut que les tissus renferment une variété de déshydrogénases spécifiques de chaque substrat : une déshydrogénase convertirait le succinate en fumarate, une autre convertirait le malate en oxalo-acétate. La déshydrogénation du succinate se faisait à une vitesse particulièrement élevée (la « succinate déshydrogénase » est l’une des quatre déshydrogénases du cycle des acides tricarboxyliques). A l’époque, une virulente controverse opposait Wieland et Warburg ; elle portait sur l’importance respective des oxydases (Warburg) ou des déshydrogénases (Wieland) dans la respiration cellulaire. Thunberg tira de ses résultats une ébauche de voie métabolique. Les atomes d’hydrogène arrachés aux substrats seraient transférés à un accepteur d’hydrogène, qui serait soit un autre métabolite, soit un accepteur artificiel comme le bleu de méthylène.
Frédéric Batelli et son assistante Lina Stern utilisèrent des broyats de tissus dans de l’eau, une solution de NaCl ou du sang, incubés dans des fioles placées un bain-marie à température constante et sous agitation mécanique. Les quantités d’O2 consommées ou de CO2 produites sont mesurées par un manomètre relié aux fioles. Ce système acellulaire permet d’étudier la respiration cellulaire, en faisant varier les paramètres. C’est ainsi qu’ils montrèrent que l’oxydation des acides organiques s’accompagne d’une production de CO2 et d’H2O. Je rappelle qu’à chaque tour de cycle des acides tricarboxyliques, deux molécules de CO2 et deux molécules de H2O sont produites.
Références : Thunberg T Bin Mikrorespirometer 1: Ein neuer Respirationsapparat, um den respiratorischen Gasaustausch kleinerer Organe und Organismen zu bestimmen (1905)
Thunberg T The Hydrogen-Activating Enzymes of the Cells (1930)
Batelli F, Stern L Recherches sur l’activité respiratoire des tissus (1907)
Batelli F, Stern L Recherches sur le mécanisme des oxydations dans les tissus des animaux isolés (1907)
Batelli F, Stern L Recherches sur les processus des combustions élémentaires dans les muscles isolés (1907)
Batelli F, Stern L Oxydation des acides malique, fumarique et citrique par les tissus animaux (1910)
Batelli F, Stern L L’oxydation de l’acide succinique comme mesure du pouvoir oxydant dans la respiration principale des tissus animaux (1910)
Cycle de Krebs
Des catalyseurs de la respiration cellulaire. Albert Szent-Györgyi von Nagyrápolt, titulaire de la chaire de Chimie médicale à l’Université de Szeged (Hongrie), utilisait la méthode manométrique mise au point par Warburg, pour mesurer la consommation en O2 d’un homogénat de muscle pectoral de pigeon ; cette consommation, d’abord élevée, diminuait avec le temps d’incubation jusqu’à cesser. S’inspirant des résultats de Torsten Thunberg, qui avait montré qu’il existe des déshydrogénases convertissant le succinate en fumarate et le malate en oxaloacétate, Szent-Györgyi constata que la consommation d’O2 était restaurée par l’addition de quantités « catalytiques » d’acides dicarboxyliques en C4 : succinate, fumarate, malate et oxaloacétate. En 1925, le neurochimiste Juda H. Quastel avait montré que le malonate, un acide dicarboxylique en C3, est un inhibiteur de la succinate-déshydrogénase. Szent-Györgyi observa que l’addition de malonate bloque la respiration cellulaire et empêche toute restauration par addition de succinate. Il proposa, en 1935, que la séquence des réactions des di-carboxylates en C4 :
Succinate – fumarate – malate – oxaloacétate
établisse un lien entre les aliments – protéines, glucides, lipides – donneurs d’hydrogène et la chaine respiratoire mitochondriale. Cette séquence de réactions est la branche des acides dicarboxyliques en C4 du futur cycle de Krebs :
succinate -2H ➔ fumarate + H2O ➔ malate -2H ➔ oxaloacétate
Références : Quastel JH Dehydrogenations Produced by Resting Bacteria. IV: A Theory of the Mechanisms of Oxidations and Reductions in vivo (1926)
Szent-Györgyi A On Oxidation, Fermentation, Vitamins, Health, and Disease (1940)
La branche des acides tricarboxyliques. En 1937, Carl Martius et Franz Knoop – le découvreur de la β-oxydation des acides gras – caractérisèrent dans le foie une déshydrogénase catalysant l’oxydation du citrate (un acide tricarboxylique en C6) en α-cétoglutarate (un dicarboxylate en C5), avec réduction de bleu de méthylène. La réaction mettait en jeu deux intermédiaires, le cis-aconitate et l’isocitrate :
citrate ➔ cis-aconitate ➔ isocitrate ➔ α-cétoglutarate
Références : Martius C, Knoop F Der physiologische Abbau der Citronensäure (1937)
Knoop F Der physiologische Abbau der Citronensäure (1937)
Hans A. Krebs avait étudié la respiration cellulaire dans le laboratoire de Otto Warburg (Kaiser Wilhelm Institut für Biologie, Berlin-Dahlem), où il s’initia à la préparation de tranches de tissus et à l’usage de la technique manométrique pour mesurer la consommation d’O2. En 1932, alors qu’il était l’assistant de Siegfried Thannhauser (Freibourg im Breisgau Universität), il avait découvert, avec son étudiant Kurt Henseleit, le cycle de l’urée, le premier cycle métabolique connu. Au cours de cette séquence de réactions, la désamination oxydative des acides α-aminés fournit deux groupes aminés qui s’unissent à une molécule de CO2 pour former une molécule d’urée (éliminée par les reins chez les animaux uréotéliques) :
NH2 – CO – NH2
Référence : Krebs H, Henseleit K Untersuchungen über die Harnstoffbildung im Tierkörper (1932)
Chassé de son poste par la politique raciale du gouvernement national-socialiste, Krebs quitta l’Allemagne en 1933 et se réfugia en Grande-Bretagne. L’appui de Sir Frederick Gowland Hopkins, prix Nobel de médecine ou physiologie 1929 et chef de file de l’école de Cambridge, lui permit de poursuivre ses recherches sur le rôle des acides carboxyliques dans la respiration cellulaire. Hans A. Krebs (The University of Sheffield) et son étudiant post-doctoral William Arthur Johnson montrèrent l’addition d’une quantité catalytique de citrate restaurait l’activité respiratoire d’un broyat de muscle pectoral de pigeon. En présence d’un inhibiteur de la succinate déshydrogénase (le malonate), il y avait accumulation d’α-cétoglutarate et de succinate.
Référence : Krebs HA, Johnson WA The role of citric acid in the intermediate metabolism in animal tissues (1937)
Krebs et Leonard V. Egglestone observèrent une accumulation de citrate lorsqu’on ajoute de l’oxalo-acétate à l’homogénat de tissus musculaire :
oxalo-acétate (C4) ➔ citrate (C6)
Krebs émit l’hypothèse que le citrate se comporte comme un catalyseur, comme l’ornithine et la citrulline (des acides aminés non protéiques) dans le cycle de l’urée. En présence de malonate, la production de succinate diminue en anaérobie ; cet inhibiteur empêche la conversion :
succinate ➔ fumarate
En aérobie, la production de succinate augmente ! L’oxalo-acétate a « chargé » un chaînon di-carboné (acétyl) donnant ainsi naissance à du citrate :
oxalo-acétate + acétyl CoA ➔ citrate
Krebs, fort de l’expérience acquise lors de ses travaux sur le Cycle de l’urée, envoya en 1937à la revue Nature un article décrivant la séquence des réactions enzymatiques du cycle qui allait porter son nom. Faisant preuve d’une myopie confondante, le comité de lecture refusa l’article ; il fut publié dans une revue nouvellement créée, Experientia. Harland G. Wood et Chester G. Werkman (Iowa State University) étudièrent le sort de l’oxalo-acétate dans le cycle de l’acide citrique. Ils incubèrent des tranches de foie de pigeon en présence de pyruvate et de bicarbonate marqué au 13C :
CH3-CO-COO- + H13CO3 ➔ -OO13C-CH2-CO-COO–
Seul le groupe carboxyle de l’oxalo-acétate adjacent au CH2 était marqué (réaction de Wood-Werkman) alors qu’on aurait pu s’attendre à un marquage des deux groupes carboxyles. Ce résultat amena Krebs à modifier la séquence des réactions du cycle ; il postula que la condensation de l’oxalo-acétate avec une molécule à 2 C issue d’un triose était un intermédiaire asymétrique en C6, le cis-aconitate, ultérieurement réduit en citrate :
oxalo-acétate ➔ cis-aconitate ➔ citrate
et en replaçant ces réactions dans l’ordre linéaire de la séquence :
➔ citrate ➔ cis-aconitate ➔ isocitrate ➔ α-cétoglutarate ➔ succinate ➔ fumarate ➔ malate ➔ oxaloacétate ➔
Références : Krebs HA, Johnson WA The role of citric acid in the intermediate metabolism in animal tissues (1937)
Krebs HA, Salvin E, Johnson WA The formation of citric and α-ketoglutaric acids in the mammalian body (1938)
Krebs HA, Egglestone LV The effect of insulin on oxidations in isolated muscle tissue (1938)
Wood HG, Werkman CH The utilization of CO2 in the dissimilation of glycerol by the propionic acid bacteria (1936)
Krebs avait d’abord appelé la voie métabolique qu’il avait découverte : « Cycle de l’acide citrique » ; tenant compte des modifications apportées à la suite des résultats de Carl Martius et Franz Knoop, il la renomma : « Cycle des acides tricarboxyliques » ; en hommage à ses découvreurs, cette voie est appelée « Cycle de Krebs » ou « Cycle de Szent Gyorgyi-Krebs ». Présent chez tous les organismes aérobies, très conservé parmi les espèces, ce cycle occupe une place centrale dans la respiration cellulaire aérobie. Localisé dans la matrice mitochondriale, les enzymes qui le composent catalysent l’oxydation d’acétyl-CoA pour générer des coenzymes réduits à haut potentiel énergétique (3 NADH, 1 FADH2) qui alimentent en électrons la chaîne respiratoire et la production d’ATP. Albert Szent-Györgyi et Hans Krebs reçurent le prix Nobel de médecine ou physiologie, le premier en 1937, le second en 1953. Une petite anecdote : Otto Warburg, jugeant que Krebs était dépourvu des qualités scientifiques requises pour faire de la recherche, lui avait refusé son appui pour obtenir un poste permanent.
J’ai évoqué au début de ce sous-chapitre l’hypothèse formulée par Albert Szent Gyorgy : la séquence des diacides carboxyliques en C4 :
succinate – fumarate – malate – oxaloacétate
établit un pont entre les aliments donneurs de H2 et la chaîne respiratoire mitochondriale. Comment se fait la transmission des e– ?
H2 ↔ 2H+ + 2e–
Considérons la manière dont l’énergie libérée par le catabolisme du glucose est récupérée ; le catabolisme se déroule dans le cytosol des cellules et chez les bactéries ; en aérobiose, le dernier catabolite du processus, le pyruvate (2 molécules par molécule de glucose), est décarboxylé en acétyle. Pour entrer dans le cycle des acides tricarboxyliques, le groupe acétyle doit être activé afin de franchir la barrière d’énergie qui s’oppose à synthèse de citrate selon la réaction décrite plus bas. L’activation se fait par association à la 4-phosphopanthétéine ; celle-ci, combinée à l’adénosine 3’,5’ diphosphate, constitue le coenzyme A (« A » pour acétylation), un transporteur de groupes acyl dans plusieurs voies métaboliques. Le CoA fut isolé en 1945 par Nathan O. Kaplan et Fritz A. Lipmann (Massachusetts General Hospital) à partir de cellules de levure et sa structure établie par James Baddiley et coll (Lister Institute of Preventive Medicine, London). La phosphopanthétéine est formée de trois molécules liées par deux liaisons de type peptidique, CO-NH : l’acide hydroxy-diméthyl-butyrique, la β-alanine, et la β-mercapto-éthylamine (cystéamine) ; le groupe thiol libre (CoA-SH) intervient dans la liaison thioester avec l’acétyle. La réaction de condensation de l’acétyl et de l’oxaloacétate a été décrite, en 1951, par Severo Ochoa (Department of Pharmacology, New York University College of Medicine) et Feodor Lynen :
acétyl-CoA (C2) + oxalo-acétate (C4) ➔ citrate (C6)
Le radical acétyle ne provient pas seulement de la dégradation du pyruvate issu du catabolisme du glucose ; la β-oxydation des acides gras et la désamination acides aminés génère aussi des fragments en C2 qui pénètrent, sous forme d’acétyl-CoA, dans le cycle de Krebs. Une partie de l’énergie libérée au cours du cycle des acides tricarboxyliques est stockée dans la liaison à haute énergie du guanosine triphosphate (GTP). Les e– à haute énergie qui sortent du cycle réduisent les coenzymes NAD+ et FAD. L’oxydation d’un radical acétyle en CO2 produit trois molécules de NADH+H+, une molécule de FADH2 et une molécule d’ATP.
Références : Kaplan NO, Lipmann FA The assay and distribution of coenzyme A (1948)
Chantrenne H, Lipmann F Coenzyme A dependence and acetyl donor function of the pyruvate-formate exchange system (1950)
Beinert H, Von Korff RW, Green DE, Buyske DA, Handschumacher RE, Higgins H, Strong FM A method for purification of coenzyme A (1952)
Chou TC, Lipmann F Separation of acetyl transfer enzymes in pigeon liver extract (1952)
Chou TC, Novelli GD, Stadtman ER, Lipmann F Fractionation of coenzyme A-dependent acetyl transfer systems (1950)
Baddiley J, Thain EM, Novelli GD, Lipmann F Structure of Coenzyme A (1953)
Stern JR, Ochoa S, Lynen F Enzymatic Synthesis of Citric Acid: V. Reaction of Acetyl Coenzyme A (1952)
Coenzymes nucléotidiques
Les êtres vivants tirent leur énergie d’électrons arrachés aux aliments au cours de réactions cataboliques. Les électrons cèdent leur énergie aux complexes de la chaîne respiratoire en passant par un intermédiaire du cycle des acides tricarboxyliques (le succinate) ou par un coenzyme nucléotidique : le diphosphopyridine nucléotide (DPN). Il fut découvert en 1906 par les biochimistes Arthur Harden et William Youndin (The Lister Institute for Preventive Medicine, London) : ils identifièrent un « co-ferment » dans des extraits de levure. Les travaux de Hans K.A.S. von Euler-Charpin et ses étudiants Ragnar Nilsson et Karl D.R. Myrbäck (Stockholms Universitet) sur la « zymase », responsable de la fermentation glycolytique chez la levure, l’amenèrent à définir le rôle et la structure nucléotidique du « phosphate de cozymase » (DPN). Rebaptisé Nicotinamide Adénine Dinucléotide (NAD) en 1961, c’est un coenzyme d’oxydoréductase actif dans des voies métaboliques comme la glycolyse ou le cycle des acides tricarboxyliques. Il a pour précurseur la vitamine B3 (niacine, vitamine PP). Hans von Euler-Charpin et Arthur Harden partagèrent le prix Nobel de chimie 1929.
Références : Harden A The Alcoholic Ferment of Yeast-Juice (1906)
von Euler H, et Ragnar Nilsson R Reinigungsversuche an Co-Zymase der Hefe (1926)
von Euler H, Myrbäck KDR Zur Kenntnis der Co-Zymase. IV. Die Konstitution des Co-Zymase (1928)
Le NAD est un accepteur ou donneur d’électrons selon la réaction :
NAD+ + H2 ↔ NADH + H+
ou, en faisant apparaître le transfert d’électrons :
NAD+ + 2H+ + 2e– ↔ NADH + H+
Lorsque la réduction du NAD+ se déroule dans la matrice mitochondriale, le NADH est oxydé par le complexe I de la chaîne des transporteurs d’électrons ; lorsqu’elle a lieu dans le cytosol, le NADH produit ne peut pas délivrer ses électrons directement à l’accepteur mitochondrial, la membrane interne des mitochondries étant imperméable au NADH. Pour franchir cette membrane, les électrons empruntent des transporteurs appelés « navettes ». Elles sont au nombre de deux : la navette du glycérol phosphate et la navette malate-aspartate. Dans le premier cas, du glycérol 3-phosphate transportant les électrons du NADH est généré par la glycérol phosphate déshydrogénase cytosolique (il existe une glycérol phosphate déshydrogénase mitochondriale dont le coenzyme est le FAD) ; les équivalents réducteurs du NADH cytosolique sont délivrés au FAD, avec réduction en FADH2 qui les délivrent à la chaîne respiratoire de la membrane mitochondriale interne. Dans le deuxième cas, la conversion d’oxalo-acétate en malate par la malate déshydrogénase cytosolique s’accompagne du chargement des électrons du NADH sur le malate. Celui-ci franchit la membrane interne. Il est reconverti en oxaloacétate par la malate déshydrogénase de la matrice mitochondriale, en délivrant les électrons au NAD mitochondrial.
En 1932, Otto H. Warburg et Walter Christian (Kaiser Wilhelm-Institut für Zellphysiologie, Berlin) isolèrent à partir de globules rouges de mammifères un enzyme catalysant l’oxydation d’un sucre-phosphate ; ils le baptisèrent : ferment jaune (das gelbe Ferment). Cet enzyme était formé d’une protéine et d’un pigment jaune, qui fut caractérisé comme étant la riboflavine 5’-phosphate (flavine mononucléotide ou FMN). La couleur jaune des flavoprotéines est dûe à l’absorption de la lumière visible par le noyau flavine. La présence de FMN fut détectée dans un complexe de la chaîne respiratoire, appelé « complexe I » ; il accepte deux électrons fournis par le NADH et les donne à l’ubiquinone par l’intermédiaire d’une protéine à Fe-S. Un autre coenzyme, apparenté au FMN, fut identifié : la flavine adénine dinucléotide (FAD) formé par l’association de FMN et d’acide adénylique. Ce coenzyme est produit sous sa forme réduite, la FADH2, au cours de deux voies métaboliques : le cycle des acides tricarboxyliques et la β-oxydation des acides gras. Au niveau du complexe II de la chaîne respiratoire, le FADH2 transfère à l’ubiquinone deux électrons fournis par le succinate. Le FAD accepte ou donne des électrons selon la réaction :
FAD + H2 ↔ FADH2
ou, en faisant apparaître le transfert d’électrons :
FAD + 2H+ + 2e– ↔ FADH2
Dans la réalité, il n’y a pas de transfert direct d’électrons d’un coenzyme à un accepteur mais « don » d’un anion hydrure H–, formé d’un proton et de deux électrons :
H– ➔ H+ + 2e–
Otto Warburg a reçu le prix Nobel de physiologie ou médecine 1931.
Références : Warburg O, Christian W Über ein neues Oxydationsferment und sein Absorptionsspektrum (1932)
Cytochromes
« La science est sujette à faire de grands sots. »
Molière
Le médecin Charles A. McMunn étudiait au spectroscope le spectre d’absorption de préparations de muscles. Mis au point en Allemagne, cet appareil optique était utilisé par les astronomes pour analyser la composition chimique des corps célestes dans le spectre de la lumière visible. Ernst F.I. Hoppe Seyler (Humbolt Universität zu Berlin) l’utilisa pour suivre les changements de spectre d’absorption de l’hémoglobine en présence d’oxyde de carbone. En 1884, McMunn observa dans les muscles d’insectes un spectre à quatre bandes d’absorption rappelant celui de l’hémoglobine mais en différant dans trois régions. Il en conclut qu’il avait découvert un nouveau pigment, qu’il baptisa « myohématine ». La poursuite de ses travaux conduisit à deux importants résultats : (i) le pigment était ubiquitaire : le spectre découvert dans les muscles d’insectes était présent dans les tissus de toutes les espèces étudiées ; le terme « myohématine » étant trop restrictif, McMunn rebaptisa le nouveau pigment : « histohématine ». (ii) Le spectre d’absorption du pigment changeait lorsque le tissu passait d’aérobiose en anaérobiose. Il émit l’hypothèse que les histohématines jouent un rôle dans la respiration cellulaire. Ces résultats, publiés en 1886, auraient dû éveiller l’intérêt des scientifiques, en particulier d’Hoppe-Seyler, pionnier de l’étude de l’hémoglobine ; c’est exactement le contraire qui se produisit. Directeur du Département de biochimie à l’Université de Strasbourg, où il instaura la Chimie physiologique comme discipline académique, fondateur et rédacteur en chef du premier journal de biochimie (Zeitschrift für Physiologische Chemie), Hoppe-Seyler déclara, sans avoir effectué la moindre vérification expérimentale, que les pigments de McMunn n’étaient que de l’hémoglobine dégradée ! En 1886, lors de la publication de ses résultats sur les histohématines, McMunn avait 34 ans et Hoppe Seyler, 61. Plutôt que de reconnaître qu’il était passé à côté d’une découverte capitale, le mandarin préféra prononcer une condamnation qui enterra pendant quatre décennies la découverte des cytochromes !
Références : McMunn CA Research on Myohematin and the Histohematins (1886)
McMunn CA Further Observations on Myohematin and the Histohematins (1886)
En 1925, l’entomologiste et parasitologiste David Keilin (The Molteno Institute, University of Cambridge) examinait au micro-spectroscope les muscles thoraciques d’un parasite du cheval (Gasterophilus intestinalis). Il observa un spectre à quatre bandes, différent de celui de l’hémoglobine. Ce spectre était observable chez d’autres parasites – Calliphora erythrocephala, Galleria mellonella – et chez la levure. De plus, lorsque la culture de levures était agitée, le spectre d’absorption apparaissait et disparaissait. Keilin arriva à la conclusion que les bandes d’absorption a (604 nanomètres), b (564 nm), c (550 nm) et d (521 nm) appartenaient à trois haemo chromogènes différents, qu’il appela « cytochromes » a, b et c. C’est en épluchant la littérature scientifique pour la publication de ses résultats que Keilin réalisa que les cytochromes avaient été découverts quarante ans plus tôt par McMunn, sous le nom d’histo hématines. Reprenant à son compte l’hypothèse de McMunn sur la fonction respiratoire des pigments, Keilin émit l’hypothèse que les cytochromes étaient des composés hématiniques servant de catalyseur redox ; au cours de la respiration cellulaire, ils étaient réduits par des déshydrogénases et oxydés par un enzyme dont le site actif contient du cuivre. Notons en passant que, devant les résultats de Keilin, Otto Warburg fit preuve du même manque de clairvoyance que Hoppe Seyler vis à vis de ceux de McMunn, en arguant que les cytochromes étaient des ferments dégénérés ne jouant aucun rôle physiologique !
Références : Keilin D On Cytochrome, a Respiratory Pigment Common to Animals, Yeasts and Higher Plants (1925)
Keilin D, Hartree F Effect of Drying upon the Absorption Spectra of Hemoglobin and its Derivatives (1952)
Keilin D Nature of the Haem-Binding Groups in native and denatured Hemoglobin and Myoglobin (1960)
Malcom Dixon (The Biochemical Laboratory, Cambridge) proposa de nommer « cytochrome oxydase » l’enzyme découvert par Otto H. Warburg (Kaiser-Wilhelm Institut für Biologie, Berlin Dahlem). David Keilin et Edward F. Hartree isolèrent le cytochrome c à partir de cœur de bœuf et montrèrent qu’il est un donneur d’électrons dans la chaîne respiratoire ; l’oxydase terminale de la respiration fut renommée : « cytochrome c oxydase ». Le Groupe de Rockefeller, George H. Hogeboom, Albert Claude, Walter C. Schneider et Rollin D. Hotchkiss (Laboratories of The Rockefeller Institute for Medical Research) montrèrent que le cytochrome c et les enzymes de la respiration cellulaire – cytochrome c oxydase, succinate déshydrogénase – sédimentent dans la fraction mitochondriale. De 1948 à 1950, Albert L. Lehninger et son graduate student Eugene P. Kennedy établirent que la chaîne des transporteurs d’électrons, le cycle de Krebs, les oxydations phosphorylantes et la β-oxydation des acides gras sont localisés dans les mitochondries.
Références : Dixon M Oxidation mechanisms in animal tissues (1929)
Keilin D, Hartree EF Purification and properties of cytochrome c (1945)
Hogeboom GH, Claude A, Hotchkiss RD The distribution of cytochrome oxidase and succinoxidase in the cytoplasm of the mammalian liver cell (1946)
Schneider WC, Claude A Hogeboom GH The distribution of cytochrome c and succinoxidase activity in rat liver fractions (1948)
Kennedy EP, Lehninger AL Oxidation of fatty acids and tricarboxylic acid intermediates by isolated rat liver mitochondria (1949)
Un lien entre respiration cellulaire et production de composés phosphorylés riches en énergie fut établi par Vladimir A. Engelhardt (Université de Kazan) : alors qu’il mesurait la consommation d’oxygène chez les globules rouges nucléés d’oiseaux, il nota que l’addition au milieu réactionnel de cyanure – un inhibiteur de la respiration cellulaire – provoque une accumulation de phosphate ; cette observation resta largement ignorée de la communauté scientifique. Engelhardt et Militza Lyubimova découvrirent que la myosine, une protéine présente dans les muscles, catalyse l’hydrolyse d’ATP en ADP et Pi en présence d’O2. Vladimir Aleksandrovich Belitser (Palladin Institute of Biochemistry, National Academy of Sciences of Ukraine, Kyiv) étudia l’influence du couple créatine/créatine-phosphate sur l’activité respiratoire de fibres musculaires. Rappelons que la créatine est une petite molécule dont la structure s’apparente à celle des acides aminés (elle en dérive) ; elle est présente dans le cerveau et les muscles sous forme libre ou phosphorylée. Des muscles de batraciens, dont la glycolyse est bloquée par addition d’un inhibiteur (iodoacétate), continuent à se contracter en consommant de la créatine phosphate. Lors d’un effort violent ou prolongé, les muscles striés épuisent leur réserve d’ATP ; ils puisent alors dans leurs réserves en esters phosphorylés – parmi lesquels la créatine phosphate – pour restaurer leur stock d’ATP. La réaction est catalysée par une kinase qui transfère le groupe phosphate de la créatine phosphate sur de l’ADP pour former de l’ATP. La créatine est phosphorylée en créatine phosphate par une autre kinase.
Référence : Engelhardt WA, Lyubimova MN Myosin and adenosine triphosphatase (1939)
La β-oxydation des acides gras
La « respiration cellulaire » est l’ensemble des processus métaboliques permettant à un être vivant de tirer des nutriments qu’il ingurgite l’énergie nécessaire à ses besoins vitaux. Dans ce contexte, les acides gras stockés dans le tissu adipeux représentent une importante source d’énergie. A titre de comparaison, l’énergie libre d’oxydation d’un acide aminé (acide glutamique) est de 478 kilocalories par molécule-gramme ; elle est de 686 pour un sucre (glucose) et de 2.338 pour un acide gras (acide palmitique). Une voie catabolique située dans la matrice mitochondriale permet l’oxydation des acides gras avec libération de cette énergie : c’est la « β-oxydation » (par référence au Cβ de la chaîne aliphatique) découverte en 1904 par le biochimiste Georg Franz Knoop (Albert Ludwig Universität Freiburg). Il donna à des chiens une alimentation contenant des acides gras saturés dont la chaîne aliphatique comportait un nombre pair ou impair d’atomes de carbone. Le dernier carbone de la chaîne – le Cω – portait un résidu aromatique – un groupe phényl. La réactivité de cette « étiquette » permit à Knoop d’identifier les produits de dégradation dans l’urine des animaux ; pour les acides ω-phénylvalérique et ω-phénylbutyrique les produits cataboliques étaient :
acide ω-phényl valérique (C4) ➔ acide benzoïque + glycine
acide ω-phényl butyrique (C3) ➔ acide phénylacétique + glycine
Knoop en tira la conclusion que la dégradation métabolique des acides gras se fait par enlèvements successifs d’une paire de C à partir de l’extrémité carboxy-terminale.
Référence : Knoop F Der Abbau aromatischer Fettsäuren im Tierkörper (1904)
Chez les êtres vivants, la plupart des acides gras ont un nombre pair d’atomes de carbone : acides laurique (C12), myristique (C14), palmitique (C16)… Ils sont synthétisés et catabolisés par addition ou élagage de composés en C2. La confirmation expérimentale de cette hypothèse buta pendant des décennies sur la difficulté à obtenir un système acellulaire fonctionnel d’oxydation les acides gras. En 1943, Luis F. Leloir et Juan M. Munoz (Fundacion Instituto Campomar, Buenos Aires) mirent au point un tel système à partir d’un homogénat de foie de rat. Ils observèrent le phénomène d’oxydation in vitro et notèrent qu’il était labile et disparaissait rapidement. Albert Lehninger (Department of Biochemistry, University of Chicago) montra qu’on pouvait le stabiliser en présence d’ATP ; il en tira la conclusion que pour être oxydés les acides gras doivent préalablement être « activés ».
Référence : Leloir LF, Munoz JM Fatty Acid Oxidation in Liver (1943)
En 1950, Feodor Lynen (Max Planck für Zellchemie, München Universität) et E. Reichart démontrèrent que l’activateur des acides gras est le coenzyme A, qui possède un groupe thiol (CoA-SH). Les acides gras sont activés dans le cytosol selon un mécanisme en deux étapes dont l’équation globale est :
acide gras + ATP + CoA-SH ➔ acyl CoA + AMP + PPi
L’acide gras activé se lie à la carnitine d’un transporteur de la membrane mitochondriale interne, la carnitine acyltransférase ; il est ainsi transféré dans la matrice mitochondriale où se déroule la dégradation des acides gras. Lynen a démontré que cette dégradation se déroule via un cycle de quatre réactions récurrentes : déshydrogénation, hydratation, déshydrogénation, et thiolyse, et identifié les intermédiaires et les enzymes impliqués. Les trois principales acyl-CoA déshydrogénases – déshydrogénases à chaîne courte (Short-chain acyl-CoA dehydrogenase), moyenne (Medium-chain acyl-CoA dehydrogenase) ou longue (Long-chain acyl-CoA dehydrogenase) – furent purifiées et caractérisées à partir de mitochondries de foie de rat par Yoshiaki Ikeda dans le laboratoire de Keiichiro Tanaka (Department of Human Genetics, Yale University School of Medicine, New Haven). Le complexe enzymatique bi-fonctionnel énoyl-coenzyme A hydratase : 3-hydroxyacyl-coenzyme A déshydrogénase a été purifié par Song-Yu Yang (Department of Pharmacology, New York State Institute for Basic Research in Developmental Disabilities, Staten Island) à partir de mitochondries de cœur de bœuf.
Références : Lynen F, Reichart E Über die Aktivierung der Essigsäure und der höheren Fettsäuren im Fettstoffwechsel (1950)
Lynen F, Ochoa S Enzymes of fatty acid metabolism (1953)
Ikeda, Y, Okamura-Ikeda K, Tanaka K Purification and characterization of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases from rat liver mitochondria. Isolation of the holo- and apoenzymes and conversion of the apoenzyme to the holoenzyme (1985)
Yang SY The large subunit of the pig heart mitochondrial membrane-bound beta-oxidation complex is a long-chain enoyl-CoA hydratase: 3-hydroxyacyl-CoA dehydrogenase bifunctional enzyme (1994)
En l’honneur de leur découvreur, on a donné aux réactions de la β-oxydation des acides gras l’appellation d’« Hélice de Lynen », une allusion au fait qu’une molécule entrant dans cette voie métabolique subit une suite de quatre réactions à la suite desquelles on revient, non pas à la molécule initiale comme dans un cycle, mais à une molécule raccourcie de deux unités C ; en effet, à chaque tour d’hélice, les acides gras à longue chaîne sont découpés en molécules à deux carbones d’acétyl-CoA avec production d’une molécule de NADH et une molécule de FADH2. Pour donner un exemple, une molécule d’acide stéarique (C18) est dégradée en huit tours de cycle en 9 molécules d’acétyl-CoA, avec production de 8 FADH2 et 8 NADH, H+. Les molécules d’acétyl-CoA seront oxydées dans le Cycle de Krebs de la matrice mitochondriale. Les coenzymes réduits, NADH, H+ et FADH2, produits dans la matrice mitochondriale par l’Hélice de Lynen et le Cycle de Krebs cèderont leurs électrons à la chaîne respiratoire de la membrane mitochondriale interne. La dégradation d’une molécule d’acide gras en C18 produit 147 molécules d’ATP, déduction faite des « frais de transport » de l’acide gras du cytosol dans la matrice mitochondriale. Lynen a reçu le prix Nobel de physiologie ou médecine 1964. En 1948 et 1949, Lehninger et Eugene Kennedy montrèrent que la β-oxydation des acides gras se déroule dans les mitochondries. L’Hélice de Lynen, comme le Cycle de Krebs, sont localisés dans la matrice mitochondriale.
Références : Lynen F, Reichart E Über die Aktivierung der Essigsäure und der höheren Fettsäuren im Fettstoffwechsel (1950)
Lynen F, Ochoa S Enzymes of fatty acid metabolism (1953)
Kennedy EP, Lehninger AL Oxidation of fatty acids and tricarboxylic acid intermediates by isolated rat liver mitochondria (1949)
Membranes associées aux mitochondries
L’existence de zones de contact entre mitochondries et reticulum endoplasmique a été rapportée en 1952 par Wilhelm Bernhard (Institut de Recherches Scientifiques sur le Cancer, Villejuif). En 1971, James D. Morré (Department of Botany and Plant Pathology, Purdue University, Lafayette, Indiana) observa l’existence de connections tubulaires, d’un diamètre de 15 à 30 nm, entre le reticulum endoplasmique lisse et la membrane mitochondriale externe des cellules animales (hépatocytes de rat) et végétales (tige d’oignon). Des zones de contacts similaires furent décrites chez les champignons par Bracker et Grove, dans le laboratoire de James Morré. Jean E. Vance (Department of Medicine University of Alberta, Edmonton) confirma l’existence de ces zones de contact en apportant une première indication sur leur rôle : il détecta une synthèse de phospholipides – phosphatidyl-sérine, phosphatidyl-éthanolamine, phosphatidyl-choline – dans une préparation de mitochondries de foie de rat. La synthèse des phospholipides se déroule dans le reticulum endoplasmique, où se déroulent les synthèses de phosphatidylcholine – par la phosphatidyléthanolamine N-méthyltransférase – et de phosphatidylsérine – par les phosphatidylsérine synthases 1 et 2. Vance eut la surprise de constater qu’une fraction mitochondriale portée à un degré de purification plus élevé avait perdu la capacité de synthétiser des phospholipides. Il en conclut que la différence entre les deux fractions mitochondriales s’expliquait par l’existence d’une « fraction X » membranaire associée à la fraction mitochondriale brute.
Note : A deux reprises, j’ai entendu mentionner l’existence de zones de contact entre mitochondries et reticulum endoplasmique sans y prêter l’attention que cela méritait. La première fois, c’était à la fin des années 1960 à l’issue d’un exposé à l’Institut de Recherches Scientifiques sur le Cancer, à Villejuif, où Wilhelm Bernhard dirigeait un laboratoire de microscopie électronique dont la réputation internationale attirait de nombreux chercheurs. Mon exposé portait sur les travaux effectués avec le Groupe de Louvain sur la caractérisation des différents types de membranes microsomiales, dont la principale est le reticulum endoplasmique. La seconde fois, c’était lors de la visite faite par James D. Morré à l’Institut de Pathologie Cellulaire et Moléculaire, à Bruxelles.
Références : Bernhard W, Haguenau F, Gautier A, Oberling C La structure submicroscopique des éléments basophiles cytoplasmiques dans le foie, le pancréas et les glandes salivaires – Étude de coupes ultrafines au microscope électronique (1952)
Vance JE Phospholipid synthesis in a membrane fraction associated with mitochondria (1990)
Morré DJ, Merritt WD, Lembi CA. Connections between mitochondria and endoplasmic reticulum in rat liver and onion stem (1971)
Bracker CE, Grove SN Continuity between cytoplasmic endomembranes and outer mitochondrial membranes in fungi (1971)
La réalité de l’existence de Membranes du reticulum endoplasmique associées aux mitochondries (Mitochondria-Associated Endoplasmic Reticulum Membranes) a définitivement été établie, principalement sur la base de deux types de résultats expérimentaux : (i) l’identification par la protéomique d’un certain nombres de protéines constitutives (Mfn2, VDAC, Grp75, PACS2) qui assurent le maintien de la stabilité de ces plateformes et participent à leur fonction ; (ii) l’observation directe de ces sites de contact par microscopie électronique à super-résolution (à l’échelle nanométrique). Selon leur origines, ces connexions ont été nommées différemment : Mitochondria Associated Membranes, MAMs, Mitochondria-ER Contacts, MERCs, chez les mammifères, ER-Mitochondria Encounter Structures, ERMES, chez la levure. Structurellement, ce sont des points de contact où la membrane mitochondriale externe et la membrane du réticulum endoplasmique sont juxtaposées – pas fusionnées – à une distance de 10 à 50 nm l’une de l’autre. Fonctionnellement, ce sont des sous-domaines spécifiques où deux organites différents, mitochondries et réticulum endoplasmique, interagissent l’un avec l’autre et coopèrent métaboliquement.
Références : Li Approaches toward super-resolution fluorescence imaging of mitochondrial proteins using PALM (2010)
Chen R, Tang X, Zhao Y, Shen Z, Zhang M, Shen Y, Li T, Ho Yin Chung C, Zhang L, Wang J, Cui B, Fei P, Guo Y, Du S, Yao S Single-frame deep-learning super-resolution microscopy for intracellular dynamics imaging (2023)
Les MAMs sont des zones de contact dynamiques, de nature protéique, entre le reticulum endoplasmique et les mitochondries ; elles occupent environ 20% de la surface de l’enveloppe mitochondriale. Ce sont des zones où s’accomplissent des transferts entre organites d’ions Ca++, de lipides, de métabolites et de molécules de signalisation. Ce sont également des zones qui participent activement à l’homéostasie du Ca++ et des lipides. Je vais, dans ce sous-chapitre, me limiter à la description des transferts de Ca++ et de lipides. Par rapport aux organites les plus volumineux de la cellule – noyau, mitochondries, chloroplastes, reticulum endoplasmique – les MAMs sont des composants mineurs ; leur purification, par les techniques classiques de fractionnement par centrifugation différentielle ou en gradient de densité est problématiques. Cela explique en partie qu’en mobilisant les ressources de la protéomique pour établir leur composition, on a identifié un grand nombre de protéines – un millier ? – dont la moitié seulement venait des mitochondries ou du réticulum endoplasmique. Il semble que la composition en protéines des MAMs varie en fonction des organes, de l’état nutritionnel ou de stress cellulaire. Un tri rigoureux des données a permis d’identifier un certain nombre de protéines de liaison qui, en établissant des interactions directes, ou en formant des complexes, assurent la stabilité de ces zones de contact. Je vais en donner un bref aperçu.
Les mitochondries ne sont pas des organites statiques ; leur activité métabolique s’accompagne d’associations transitoires en structures ramifiées. En relation avec ces phénomènes de fusions et de scissions, Yuka Eura, Naotada Ishihara et coll. (Department of Molecular Biology, Graduate School of Medical Science, Kyushu University, Fukuoka) ont caractérisé, chez le rat, deux protéines isoformes : les mitofusines Mfn1 et Mfn2. Ces deux protéines présentent des analogies de séquence avec la GTPase transmembranaire Fzo (FuZzi Onion) impliquée, chez la Drosophile, dans la fusion des mitochondries entre elles. L’observation par immuno-microscopie électronique d’un mutant de Mfn2 a révélé que cette protéine se concentre aux points de contact entre mitochondries sur le point de fusionner. Mfn1induit la formation de structures tubulaires en réseau. Les résultats d’expériences complémentaires ont établi que, dans les cellules de mammifères, les deux mitofusines coopèrent dans la fusion des mitochondries. Ishihara, Eura et coll. ont comparé, par microscopie à fluorescence, l’importance de l’ancrage (tethering) dépendant du GTP chez des mitochondries exprimant soit Mfn1 soit Mfn2. Pour différencier les deux types de protéines, les auteurs ont marqué les mitofusines avec des protéines fluorescentes vertes ou rouges. Ils sont arrivés à la conclusion qu’elles ont des rôles différents et que seul Mfn1, en présence de GTP, est responsable de l’ancrage (tethering) mitochondries-reticulum.
Références : Eura Y, Ishihara N, Yokota S, Mihara K Two mitofusin proteins, mammalian homologues of fzo, with distinct functions are both required for mitochondrial fusion (2003)
Ishihara N, Eura Y, Mihara K Mitofusin 1 and 2 play distinct roles in mitochondrial fusionreactions via GTPase activity (2004)
Les mitofusines appartiennent à la famille des « dynamin-related GTPases ». Pour rappel, les dynamines – il en existe une variété – sont des GTPases pouvant s’auto-assembler en hélices ; elles jouent un rôle essentiel dans le transport vésiculaire entre organites ou dans l’endocytose. Les mitofusines sont des protéines intégrales de la membrane mitochondriale externe (Mfn1) ou de la membrane du reticulum endoplasmique (Mfn2). Si le rôle de Mfn1 a été rapidement établi, celui de Mfn2 – attache (tether) ou espaceur (spacer) ? – fut plus laborieux à définir. Olga Martins de Brito et Luca Scorano (Dulbecco-Telethon Institute, Venetian Institute of Molecular Medicine, Padova) ont montré: (i) que Mfn2 est concentré à l’interface entre mitochondries et reticulum endoplasmique ; (ii) que chez les fibroblastes d’embryon de souris ou dans les cellules HeLa, l’absence de Mfn2 provoque une dislocation du reticulum endoplasmique et des plateformes de contact avec les mitochondries, ce qui a pour effet de diminuer le transfert d’ions Ca++ dans les mitochondries. Sur la base des résultats d’expériences biochimiques et génétiques in vitro, Brito et Scorano ont proposé un modèle dans lequel la Mfn2 du reticulum participe à l’ancrage des mitochondries en formant des interactions homotypiques Mfn2 – Mfn2 ou hétérotypiques Mfn2 – Mfn1. Cependant, la question du rôle de Mfn2 a encore été débattue. Nuno Santos Leal et coll. (Center for Alzheimer Research, Department of Neurobiology, Karolinska Institutet, Stockholm) ont montré que le « silençage » du gène de Mfn2 par un petit ARN interférent (siRNA knockdown) augmente le nombre de points de contact entre mitochondries et reticulum endoplasmique ainsi que la longueur des zones de contact, avec augmentation du transfert de Ca++ dans les mitochondries. Domenico Cieri et coll. (Department of Biology, University of Padova) ont montré que l’inhibition de Mfn2 influe sur la structure des MAMs : la proportion de MAMs « étroites » augmente et celle de MAMs « larges » diminue.
Références : de Brito OM, Scorrano L Mitofusin 2 tethers endoplasmic reticulum to mitochondria (2008)
Leal NS, Schreiner B, Pinho CM, Filadi R, Wiehager B, Karlström H, Pizzo P, Ankarcrona M Mitofusin-2 knockdown increases ER–mitochondria contact and decreases amyloid β-peptide production (2016)
Cieri D, Vicario M, Giacomello M, Vallese F, Filadi R, Wagner T, Pozzan T, Pizzo P, Scorrano L, Brini M, Cali T SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition (2017)
Cosson P, Marchetti A, Ravazzola M, Orci L Mitofusin-2 Independent Juxtaposition of Endoplasmic Reticulum and Mitochondria: An Ultrastructural Study (2012)
Ainbinder A, Boncompagni S, Protasi F, Dirksen RT Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle (2015)
Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling (2015)
Note : De nombreuses publications sont consacrées aux mitofusines, en particulier dans le cadre de leur implications dans un certain nombre de pathologies. Dans le cadre d’un sous-chapitre consacré aux Membranes associées aux mitochondries, je me limiterai au texte ci-dessus.
L’adhésion cellulaire au sein d’un tissu est assurée par un certain nombre de jonctions ; les protéines de la famille des cadhérines en sont des constituants essentiels. Il s’agit de glycoprotéines membranaires Ca++-dépendantes (d’où leur nom), de grande taille (120 – 140 kDa), localisées dans la membrane plasmique. Leur extrémité carboxy-terminale entre en interaction avec le cytosquelette d’actine par l’intermédiaire des caténines. Cette association est régulée par la phosphorylation de résidus tyrosine. Janne Balsamo et coll. (Department of Biological Sciences, Wayne State University, Detroit) ont cloné une phosphatase de protéine-tyrosine (Protein Tyrosine Phosphatase) nommée PTP1B, associée au domaine cytoplasmique des cadhérines, et qui régule le rôle des N-cadhérines dans l’adhésion cellulaire.
Références : Balsamo J, Arregui C, Leung T, Lilien J The nonreceptor protein tyrosine phosphatase PTP1B binds to the cytoplasmic domain of N-cadherin and regulates the cadherin-actin linkage (1998)
Xu G, Arregui C, Lilien J, Balsamo J PTP1B modulates the association of beta-catenin with N-cadherin through binding to an adjacent and partially overlapping target site (2002)
Les phosphatases de protéines-tyrosine sont, avec les protéines kinases, des acteurs importants de la signalisation cellulaire et de la régulation des fonctions métaboliques. Elles sont principalement localisées dans le cytosol, le noyau ou la membrane plasmique. En 1999, Stöger et coll. ont montré que PTP1B, un nouveau membre de la famille des phosphatases de protéine-tyrosine, est localisée dans les mitochondries, ce qui suggère que cette phosphatase joue un rôle dans un mécanisme de régulation des fonctions mitochondriales par signalisation tyrosine. En utilisant le système de double hybride basé sur l’emploi du facteur de transcription LexA, Porsche A. Allensbach, doctorant à l’Université de Constance, a mis en évidence l’interaction in vitro entre une protéine transmembranaire et les phosphatases de protéine-tyrosine PTP1B et TcPTP (T-cell protein tyrosine phosphatase). Cette protéine transmembranaire, de la famille des protéines RMDN (Regulator of Microtubule DyNamics), fut nommée PTPIP51 (Protein Tyrosine Phosphatase-Interacting Protein 51).
Références : Stöger C, O’Connor V, Miller CC, Ackerley S, Lau KF, Miller JB Human and mouse PTPB (protein tyrosine phosphatase-like protein) 51 is a novel mitochondrial protein tyrosine phosphatase (1999)
Porsche A. Allensbach Identifikation von Interaktion-spartnern der T-Zell Protein-Tyrosin-Phosphatase durch das Lex-A Two Hybrid System thesis (2001)
Albrecht Stenzinger et coll. (Institute of Anatomy and Cell Biology, Justus-Liebig-University Giessen) ont mené une vaste étude sur la distribution de PTPIP51 (ARNm et protéine) dans les organes du cobaye, du rat, de la souris, du porc et de l’homme. Leurs résultats suggèrent que PTPIP51 est impliquée dans les mécanismes de régulation de la différentiation, du mouvement des cellules, ou de l’organisation du cytosquelette. Sa localisation dans les mitochondries des cellules d’origine humaine HEK293T et HeLa fut établie par Bingfeng Lv et coll. (Laboratory of Medical Immunology, School of Basic Medical Science, Peking University Health Science Center), au cours de l’étude qu’ils menaient sur le mécanisme de l’apoptose, dans lequel les mitochondries jouent un rôle central. Ils montrèrent que PTPIP51 est une protéine transmembranaire et que son adressage dans les mitochondries est dirigé par un signal situé à proximité de son extrémité N-terminale. PTPIP51 – en particulier son domaine N-terminal – pourrait être impliquée dans la voie de l’apoptose médiée par le cytochrome c mitochondrial.
Références : Stenzinger A, Kajosch T, Tag C, Porsche A, Welte I, Hofer HW, Steger K, Wimmer M The novel protein PTPIP51 exhibits tissue- and cell-specific expression (2005)
Lv BF, Yu CF, Chen YY, Lu Y, Guo JH, Song QS, Ma DL, Shi TP, Wang L Protein tyrosine phosphatase interacting protein 51 (PTPIP51) is a novel mitochondria protein with an N-terminal mitochondrial targeting sequence and induces apoptosis (2006)
Les protéines de la famille VAP (Vesicle-Associated membrane Proteins) sont des protéines homologues présentes chez l’homme, la drosophile et Cenorhabditis elegans. C’est par leur interaction avec la synaptobrévine membranaire des vésicules synaptiques, qu’elles ont été identifiées, d’où les appellations : Synaptobrevine/VAP Vesicle-Associated Membrane Protein ou VAMP-Associated Protein. Elles possèdent un domaine transmembranaire – inséré dans la membrane du reticulum endoplasmique pour VABP – et un domaine amino-terminal MSP (Major Sperm Protein) ; ce motif est analogue aux structures en sandwich des immunoglobulines – deux feuillets β antiparallèles. Les protéines VAP interagissent par leur domaine N-terminal avec lesmicrotubules et interviennent dans la motilité, le transport membranaire, la signalisation extracellulaire et différentes interactions protéine-protéine. Leur étude ne suscitait qu’un intérêt modéré jusqu’au jour où Agnes L. Nishimura et coll. (Human Genome Research Center, Department of Biology, Biosciences Institute, São Paulo University) et d’autres groupes ont démontré l’implication de l’une d’entre elle, VABPB (Vesicle-Associated membrane Protein associated protein B), dans les maladies affectant les neurones moteurs qui transmettent aux muscles les signaux venant du cortex cérébral. L’une des mieux connues et des plus anciennement étudiées de ces pathologies est la maladie décrite en 1869 par Jean-Martin Charcot (Hôpital de la Salpêtrière, Paris) : la sclérose latérale amyotrophique.
Références : Nishimura AL, Mitne-Neto M, Silva HCA, Richieri-Costa A, Middlleton S, Dulio C, Kok F, Oliveira JRM, Gillingwater T, Webb, J, Skehel P, Zatz M A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis (2004)
Blair K, Martinez-Serra R, Gosset P, Martín-Guerrero SM, Mórotz GM, Atherton J, Mitchell JC, Markovinovic A, Miller CCJ Structural and functional studies of the VAPB-PTPIP51 ER-mitochondria tethering proteins in neurodegenerative diseases (2025)
Aux sites de contact MAM (Mitochondria-Associated Membranes), la protéine PTPIP51 (Protein Tyrosine Phosphatase Interacting Protein 51), de la membrane mitochondriale externe, et la protéine VAPB (Vesicle-Associated Membrane Protein B), de la membrane du reticulum endoplasmique, interagissent pour former un complexe d’attache (tether) entre ces deux membranes ; cette attache est aussi un élément de la stabilité des MAM. La formation du complexe d’attache se fait par interaction directe et spécifique entre deux domaines des protéines impliquées : le domaine MSP de VAPB lie directement le motif cytosolique FFAT-like de PTPIP51, riche en phénylalanine et en tyrosine. Le domaine coiled-coil de PTPIP51 participerait aussi à l’interaction. La communication entre les deux organites est établie quand la distance séparant les deux membranes est de l’ordre de 10 à 80 nm. Kurt J. De Vos et coll. (Department of Neuroscience, MRC Centre for Neurodegeneration Research, Institute of Psychiatry, King’s College London) ont démontré l’importance de l’interaction PTPIP51 – VABP dans l’homéostasie du Ca++.
Références : De Vos K, Mórotz GM, Stoica R, Tudor EL VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis (2011)
De Vos K, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CCJ VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis (2012)
Je vais décrire un autre complexe protéique de contact dans le sous-chapitre consacré à l’homéostasie du calcium.
Homéostasie du calcium
Le réticulum endoplasmique est l’organite central du métabolisme du Ca++ et de son homéostasie cellulaire ; il est le site de stockage de ce cation essentiel. La concentration en Ca++ du cytosol est de l’ordre de la nanomole ; il atteint un niveau voisin de la micromole dans le lumen du reticulum. Le Ca++ est un facteur essentiel de la signalisation cellulaire ; il entre et sort continuellement du reticulum ; sa concentration active dans le lumen ou le sarcoplasme est assurée par les ATPases Ca++ (Sarcoplasmic/Endoplasmic Reticulum Ca2+-ATPase, SERCA). Ces pompes transmembranaires ont été découvertes dans le laboratoire de Fritz A. Lipmann (Rockefeller Institute, New York). Les SERCA maintiennent la concentration cytosolique au niveau nanomolaire. Dans le lumen, le Ca++ est lié coopérativement à la calséquestrine, une protéine découverte, isolée et purifiée par David H. MacLennan et Peter R. Wong (Banting and Best Department of Medical Research, University of Toronto). Cette protéine acide, dotée de « mains EF (EF hand) », peut capturer jusqu’à 50 Ca++ par molécule. Le motif EF consiste en deux hélices α reliées par une boucle d’une douzaine d’acides aminés ; chaque main peut chélater un ion Ca++.
Note : Fritz A. Lipmann, le découvreur du coenzyme A, a partagé avec Hans A. Krebs le prix Nobel de physiologie ou médecine 1953.
Références : Ebashi S, Lipmann F Adenosine-Triphosphate-linked Concentration of Calcium Ions in a Particulate Fraction of Rabbit Muscle (1962)
Hasselbach W ATP-driven active transport of calcium in the membranes of the sarcoplasmic reticulum (1964)
MacLennan DH, Wong PRS Isolation of an Acidic Protein from Rabbit Skeletal Muscle Sarcoplasmic Reticulum Which Binds Calcium and Is Released by High Salt Concentration (1971)
Lorsque le besoin de Ca++ dans le cytosol se fait sentir, le stock accumulé dans le lumen est libéré par des canaux ioniques transmembranaires du reticulum endoplasmique. Katsuhiko Mikoshiba et son équipe (Laboratory of Cell Calcium Signaling, Shanghai Institute for Advanced Immunochemical Studies) ont identifié ces canaux en montrant que la protéine P400, présente dans le cervelet, est en réalité le récepteur de l’Inositol-1,4,5-triphosphate ; on en a identifié trois types : IP3R1, IP3R2, IP3R3. Leur activité est régulée par l’inositol triphosphate, un messager intracellulaire, présent dans le cytosol ; c’est le produit d’hydrolyse, par la phospholipase C, d’un phospholipide de la membrane plasmique des cellules eucaryotes, le phosphatidylinositol 4,5-bisphosphate. Le récepteur à IP3 a été purifié à partir de cerveaux de rats par Solomon Snyder et son équipe (Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore). Gregory A. Mignery et coll. (Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas) ont démontré la similarité de structure entre le récepteur de l’IP3 et le récepteur de la ryanodine – un alcaloïde d’origine végétale – du reticulum sarcoplasmique des fibres musculaires. Dans le cytosol, les ions Ca++ sont pris en charge par la calmoduline (Calcium-modulated Protein, CaM). Cette protéine de petite taille (148 résidus d’acides aminés) a été découverte par Wai Yiu Cheung au cours d’une étude sur la phosphodiestérase de l’AMP 3′,5′ cyclique dans le cerveau de bœuf et le venin de serpent ; cet enzyme ne fonctionne qu’en présence d’un activateur. Shiro Kakiuchi et son équipe arrivèrent à la même conclusion et à la dépendance de la phosphodiestérase au Ca++. Wai Yiu Cheung proposa de nommer l’activateur de la phosphodiestérase : calmodulin (CALcium MODULated proteIN). En changeant de conformation après chélation d’un atome de Ca++ par l’une de ses quatre mains EF, la calmoduline se comporte comme un senseur du Ca++ intra-cellulaire. Le complexe Ca++– calmoduline régule l’activité de kinases sérine-thréonine-dépendantes, les protéines kinases Ca++/calmoduline dépendantes qui interviennent dans la contraction musculaire et la libération de neurotransmetteurs par le système nerveux.
Références : Furuichi T, Yoshikawa S, Miyawaki A, Wada K, Maeda N, Mikoshiba K Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400 (1989)
Supattapone S, Worley PF, Baraban J M, Snyder SH Solubilization, purification, and characterization of an inositol trisphosphate receptor (1988)
Mignery GA, Südhof TC, Takei K, De Camilli P Putative receptor for inositol 1,4,5-trisphosphate similar to ryanodine receptor (1989)
Cheung WY Cyclic 3′,5′-nucleotide phosphodiesterase. Demonstration of an activator (1970)
Kakiuchi S, Yamazaki R Calcium dependent phosphodiesterase activity and its activating factor (PAF) from brain studies on cyclic 3′,5′-nucleotide phosphodiesterase (1970)
Article de revue : Stokes DL, Wagenknecht T Calcium transport across the sarcoplasmic reticulum: structure and function of Ca2+-ATPase and the ryanodine receptor (2000)
Les besoins en Ca++ de la mitochondrie sont constants. Pour les satisfaire, le Ca++ stocké dans le lumen du réticulum endoplasmique est importé dans les mitochondries au niveau de la zone de contact des MAMs. Ce transfert se fait par une série de canaux faisant partie d’un complexe macromoléculaire : le premier canal est le récepteur de l’inositol triphosphate inséré dans la membrane du réticulum ; son ouverture est commandée par la liaison du ligand, l’inositol triphosphate cytosolique ; au niveau des MAMs, le domaine du récepteur qui émerge dans le cytosol lie une chaperonne cytosolique : la GRP75 (Glucose-Regulated Protein 75) qui, elle-même, se lie à un canal inséré dans la membrane mitochondriale externe : le canal VDAC (Voltage-Dependent Anion Channel). Le pont moléculaire établi par la GRP75 permet au Ca++, qui a traversé successivement les deux canaux du complexe VDAC-GRP75-IP3R, de pénétrer dans l’espace inter-membranaire mitochondrial. Pour franchir la membrane mitochondriale interne et parvenir dans la matrice mitochondriale – où se déroulent le cycle de Krebs et la synthèse d’ATP – le Ca++ traverse un troisième canal transmembranaire : le complexe de l’Uniporteur de calcium mitochondrial (Mitochondrial Calcium Uniporter) ; le potentiel négatif de la membrane mitochondriale interne favorise le transfert électrophorétique du Ca++.
Référence : De Stefani D, Patron M, Rizzuto R Structure and function of the Mitochondrial Calcium Uniporter complex (2015)
Homéostasie des lipides
En 1952, Arthur Kornberg et W.E. Pricer junior (Department of Microbiology, Washington University, Saint Louis) montrèrent que des enzymes présents dans des fractions subcellulaires de foie catalysent, in vitro, l’incorporation de phosphocholine marquée au 32P, en présence d’ATP, dans un lipide, qu’ils identifièrent ultérieurement à la lécithine. Ils proposèrent la réaction suivante :
Phosphatidic acid + [32P] phosphocholine ➔ [32P] phosphatidylcholine + Pi
Référence : Kornberg A, Pricer W E Jr Enzymatic Synthesis of Phosphorus-Containing Lipides (1952)
Eugene P. Kennedy (Department of Biochemistry, University of Chicago) s’était fixé pour but d’élucider le mécanisme de formation de la liaison phosphodiester de la lécithine (phosphatidylcholine).. Dans le laboratoire d’Albert Lehninger (Department of Biochemistry, University of Chicago), Kennedy, encore étudiant, avait eu l’occasion de préparer des mitochondries par centrifugation dans le saccharose 0,88M, la méthode mise au point par George Palade au Rockefeller Institute. Il adopta une approche biochimique in vitro en utilisant une fraction subcellulaire de foie de rat – dont le composant principal était des mitochondries – et le double marquage au 32P et au 14C pour suivre le sort de la choline dans les produits de réaction. Il découvrit que la 14C-choline – mais pas la 32P-phosphocholine – était incorporée dans une fraction lipidique qu’il identifia à la lécithine (phosphatidylcholine) ; l’incorporation était stimulée par l’addition de coenzyme A et d’AMP, que les phosphorylations oxydatives mitochondriales transformaient en ATP. Dans des expériences ultérieures, il identifia et isola les enzymes impliqués dans l’incorporation de la choline dans la lécithine : la choline kinase et la CTP: phosphocholine cytidylyltransférase.
Référence : Kennedy EP The Synthesis of Lecithin in Isolated Mitochondria (1953)
En 1954, Clark Bublitz et Eugene Kennedy identifièrent, dans une fraction mitochondriale de foie de rat, l’enzyme qui catalyse la phosphorylation du glycérol – la réaction initiale de la biosynthèse de la lécithine :
Glycérol kinase + Mg2+
Glycérol + ATP ➔ L-α-Glycérophosphate + ADP
Le glycérol-3-phosphate provient du glycérol ou de la dihydroxyacétone phosphate ; son estérification par un premier acide gras conduit à l’acide lysophosphatidique, puis par un second acide gras, à l’acide phosphatidique, le précurseur commun des triglycérides et des glycérophospholipides.
Référence : Bublitz C, Kennedy EP Synthesis of phosphatides in isolated mitochondria. III. The enzymatic phosphorylation of glycerol (1954)
Les résultats de Kennedy et ceux de Kornberg & Pricer divergeaient sur un point important : la possibilité ou pas d’incorporer de la phosphocholine dans un lipide. Avant de comparer les conditions expérimentales utilisées par les deux groupes Samuel B. Weiss, un étudiant post-doctoral, commença par vérifier que le système d’incubation in vitro utilisé par Kennedy générait bien de l’ATP à partir d’AMP. Le fait que ce nucléotide est le produit des phosphorylation oxydatives ne faisait que renforcer Kennedy dans sa conviction que la biosynthèse des phospholipides se déroule dans les mitochondries. Le Groupe de Louvain a démontré que les mitochondries, si elles sont le composant principal des fractions mitochondriales préparées par centrifugation, elles ne sont pas le seul. Reprenant les conditions expérimentales de Kornberg & Pricer, Weiss remplaça l’ATP produit in vitro par une préparation commerciale ; il eut la surprise de constater qu’il pouvait alors reproduire leur résultat : la phosphocholine était incorporée dans un la lécithine à la condition d’ajouter des quantités importantes d’ATP. Par contre, avec une préparation cristallisée d’ATP, il n’y avait pas d’incorporation. Une conclusion s’imposait : le facteurs stimulant l’incorporation n’était pas l’ATP mais un contaminant ; plutôt que d’essayer de l’identifier au prix d’une laborieuse purification, Weiss et Kennedy essayèrent d’autres nucléotides : le contaminant actif était la cytidine triphosphate. Ils émirent l’hypothèse que la cytidine diphosphocholine est un intermédiaire à haute énergie de la synthèse des lipides. Pour le prouver, ils isolèrent et caractérisèrent la CDP-choline et la CDP-éthanolamine à partir de foie de rat et de levure. Après marquage au 14C, ils montrèrent qu’elles sont les précurseurs activés de la phosphorylcholine et de la phosphoryléthanolamine dans la synthèse de la lécithine et de la phosphatidyléthanolamine, les phospholipides membranaires les plus abondants chez les mammifères.
Référence : Kennedy EP, Weiss SB The function of cytidine coenzymes in the biosynthesis of phospholipides (1956)
Avant les travaux de Kennedy, le processus exact par lequel les acides gras, le glycérol et les bases azotées (comme la choline) étaient assemblés pour former des phospholipides restait un mystère. Dans un article publié en 1956, Kennedy a proposé une séquence des réactions enzymatiques de la synthèse de phosphatidylcholine et de phosphatidyléthanolamine. En hommage à leur découvreur, on les appelle « Voie de Kennedy pour la synthèse des phospholipides » (Kennedy Pathway for phospholipid biosynthesis) ou voie CDP-choline/CDP-éthanolamine. Le tableau suivant en donne un résumé :
Choline kinase
choline + ATP ➔ phosphocholine + ADP
CDP-choline synthase
phosphocholine + CTP ➔ CDP-choline + PPi
CTP: phosphocholine cytidyltransférase
CDP-choline + diacylglycérol ➔ phosphatidylcholine + CMP
Voie de Kennedy. La CTP: phosphocholine cytidyl transferase
est l’enzyme régulateur de l’étape limitante de la synthèse de
la phosphatidylcholine
La synthèse de la phosphatidyléthanolamine se déroule selon un mécanisme similaire, en trois étapes, catalysées par l’éthanolamine kinase, la CTP : éthanolamine-phospho cytidyl transférase et l’éthanolamine-phosphotransférase.
Référence : Kennedy EP The Biological Synthesis of Pospholipids (1956)
Reticulum endoplasmique et mitochondries des hépatocytes sont les deux organites au centre de l’homéostasie lipidique. L’essentiel des lipides de la cellule – triglycérides, phospholipides, stéroïdes – sont synthétisés par des enzymes situés sur la face cytosolique du domaine lisse du reticulum endoplasmique ; il est le seul organite dans lequel s’effectue la synthèse de la phosphatidylcholine, des sphingolipides et du cholestérol. Les mitochondries ne synthétisent que certains lipides, comme la cardiolipine et les stéroïdes. Le renouvellement constant de leurs phospholipides membranaires, particulièrement de ceux de la membrane interne où se déroule la synthèse d’ATP, est une nécessité pour le maintien de leur intégrité fonctionnelle ; il dépend d’un transfert constant de phospholipides à partir du reticulum endoplasmique. Les Membranes du reticulum endoplasmique associées aux mitochondries (MAMs) sont le site de ces échanges. Les complexes PTPIP51-VAPB exercent une double fonction : (i) ils servent d’attaches (tethers) pour maintenir les deux membranes à proximité l’une de l’autre ; (ii) ils captent et transfèrent des phospholipides parmi lesquels l’acide phosphatidique, un précurseur dans la synthèse de la cardiolipine de la membrane mitochondriale interne. Dans les cellules déficientes en PTPIP51, on observe des teneurs réduites en cardiolipine mitochondriale.
D’autres protéines contribuent à la diffusion passive des phospholipides vers les mitochondries : les Protéines de transfert lipidique (Lipid Transfer Proteins) sont des tunnels hydrophobes entre les membranes du reticulum et des mitochondries. Ce sont de longues protéines, en forme de tiges, dotées d’un canal hydrophobe interne (Rainure Hydrophobe) dans lequel s’insèrent les queues hydrophobes des acides gras des phospholipides, tandis que les tête polaires sont stabilisées. Les lipides, ainsi préservés du contact avec la phase aqueuse, coulissent d’une membrane à l’autre en empruntant le tunnel. Un certain nombre de ces les Protéines de transfert lipidique ont été caractérisées : les Multiple Co-enzyme A binding proteins, les protéines interagissant avec la choline (dites à domaine choline) : la protéine Vps13 de Saccharomyces cerevisiae et la protéine d’autophagie Atg2. Toutes sont de longues molécules établissant des ponts entre les membranes de deux organites et s’associant pour former un canal hydrophobe. Chez la levure, le transfert de l’acide phosphatidique est assuré par les protéines – Mmm1, Mdm10, Mdm12, Mdm34 – du complexe ERMES (ER-Mitochondria Encounter Structure).
Références : Engel C, Wieringa R The diverse world of coenzyme A binding proteins (1996)
Ugur B, Hancock-Cerutti W, Leonzino M, De Camilli P Role of VPS13, a protein with similarity to ATG2, in physiology and disease (2020)
Dziurdzik SK, Conibear E The Vps13 Family of Lipid Transporters and Its Role at Membrane Contact Sites (2021)
ATP et respiration cellulaire
Dans les années 1930 et 1940, la mise en évidence du lien entre respiration cellulaire et production d’énergie – sous forme d’ATP – fut une avancée majeure. J’en rappelle brièvement les étapes : en 1929, H. Karl A.H. Lohmann, assistant du biochimiste Otto F. Meyerhof (Kaiser Wilhelm Institute für Biologie, Berlin-Dahlem, prix Nobel de physiologie ou médecine 1922), découvrit l’adénosine triphosphate (ATP). Il crut d’abord que ce nucléotide était spécifique des muscles ; des études ultérieures montrèrent que cette molécule était présente dans toutes les cellules et qu’elle intervenait dans pratiquement tous les processus cellulaires nécessitant un apport d’énergie. Il fut établi que de l’ATP était synthétisé au cours de la glycolyse anaérobie. Cette voie – la première voie métabolique élucidée – ne représente pas chez les cellules eucaryotes, qui vivent en aérobiose, un moyen efficace de produire de l’énergie : les trois premières réactions de la glycolyse consomment autant de molécules d’ATP qu’elles en produise (deux).
Référence : Lohmann K The pyrophosphate fraction in muscle (1929)
J’ai mentionné plus haut que l’emploi de la coloration au Vert Janus de Leonor Michaelis avait révélé l’existence de réactions d’oxydoréduction au sein des mitochondries. En 1933, Herman M. Kalckar commença une thèse de doctorat dans le laboratoire de physiologie d’Ejnar Lundsgaard, à l’université de Copenhague, sous la direction de Fritz A. Lipman, récemment émigré d’Allemagne. En 1937, Kalckar montra que dans un milieu réactionnel contenant un extrait acellulaire de cortex rénal et du fluorure de sodium (pour inhiber la glycolyse anaérobie et les phosphatases cellulaires susceptibles de détruire l’ATP), l’addition d’intermédiaires du cycle des acides tricarboxyliques provoquait une synthèse d’ATP avec réduction d’oxygène. La réaction ne se produisait pas en anaérobiose ou en présence de cyanure (un inhibiteur de la respiration cellulaire). Le lien entre respiration cellulaire et synthèse d’ATP fut confirmé par les résultats des travaux de V.A. Belitzer et E.T. Tsybakova (Instytut Biokhimyi, Kiev), qui montrèrent que l’oxydation de certains substrats ne se déroulait qu’en présence d’ADP et de phosphate inorganique, et par ceux de Severo Ochoa (Washington University, St Louis, prix Nobel de physiologie ou médecine 1959), qui étudia le métabolisme de la créatine dans le laboratoire de D. Noël Paton, à Glasgow, et chez Otto Meyerhof. Kalckar et Lipman, indépendamment l’un de l’autre, créèrent le concept de « liaison riche en énergie » par référence au caractère très exergonique de de la liaison anhydride d’acide phosphorique de l’ATP ; dans les conditions prévalant dans le milieu cellulaire, la variation d’enthalpie libre lors de l’hydrolyse de l’ATP en ADP vaut :
ΔG = – 51,8 kJ/mole
Plutôt que le joule – appartenant au Système International d’Unités -, les biochimistes préfèrent exprimer l’énergie en calories, une unité introduite en 1824 par le physico-chimiste Nicolas Clément. Le transfert du groupe phosphate terminal de l’ATP s’accompagne de la libération d’une énergie libre standard d’hydrolyse de 7,3 kilocalories par mole.
Références : Kalckar HM Phosphorylation in Kidney Tissue (1937)
Belitzer VA, Tsybakova ET Biokhimiya On the Respiration and Phosphorylation of Muscle Tissue (1941)
Ochoa S ”Coupling” of Phosphorylation with Oxidation of Pyruvic Acid in Brain Tissue (1940)
Malgré cette richesse en énergie chimique, et contrairement à ce qui est parfois enseigné dans les manuels de biochimie, l’ATP n’est pas une forme de stockage de l’énergie ; son existence est trop brève, sa demi-vie dans la cellule étant de l’ordre de la minute. Il faut plutôt considérer l’ATP comme le transporteur universel d’énergie et le couple ADP/ATP, comme une batterie chimique constamment rechargée par phosphorylation. En 1941, Fritz A. Lipmann (Massachusetts General Hospital, Boston, prix Nobel de médecine ou physiologie 1953), le découvreur du coenzyme A, démontra le rôle central joué par l’ATP dans les transferts d’énergie chimique au sein de la cellule. On estime que la consommation d’un individu en ATP par 24 heures est équivalente à sa masse corporelle.
Mettant à profit l’avancée technologique du fractionnement subcellulaire quantitatif par centrifugation différentielle, codifié par Albert Claude (Rockefeller Institute), un groupe de chercheurs incluant Walter C. Schneider et Van Rensselaer Potter (McArdle Laboratory for Cancer Research, University of Wisconsin, Madison), George H. Hogeboom et Rollin D. Hotchkiss (National Institutes of Health, Bethesda) montrèrent que le cytochrome c et les enzymes de la respiration cellulaire (cytochrome oxydase, succinate déshydrogénase) et de l’oxydation des acides gras, sédimentent dans la fraction mitochondriale. Albert L. Lehninger et son graduate student Eugene P. Kennedy (University of Chicago) observèrent que, dans une fraction subcellulaire de foie de rat, les phosphorylations oxydatives et la β-oxydation des acides gras sont localisés dans un même organite entouré d’une membrane, imperméable à certains soluté. Ils émirent l’hypothèse que cet organite était la mitochondrie, où se déroule le Cycle de Krebs.
Références : Claude A Fractionation of mammalian liver cells by differential centrifugation I. Problems, methods, and preparation of extract (1946)
Claude A Fractionation of mammalian liver cells by differential centrifugation II. Experimental procedures and results (1946)
Potter VR The assay of animal tissues for respiratory enzymes; cell structure in relation to fatty acid oxidation (1946)
Hogeboom GH, Claude A, Hotchkiss RD The distribution of cytochrome oxidase and succinoxidase in the cytoplasm of the mammalian liver cell (1946)
Schneider WC, Claude A, Hogeboom GH The distribution of cytochrome c and succinoxidase in rat liver fractions (1948)
Kennedy EP, Lehninger AL Oxidation of fatty acids and tricarboxylic acid cycle intermediates by isolated rat liver mitochondria (1949)
Potter VR, Lyle GG, Schneider WC Oxidative phosphorylation in whole homogenates and in cell particles (1951)
Des électrons en cascades
Avec ses quatre compartiments (membrane externe, espace inter-membranaire, membrane interne, matrice) associés à des fonctions différentes, la mitochondrie est un organite complexe. La membrane interne est le site d’un transport d’électrons et de production d’énergie chimique. Cette « chaîne » respiratoire fournit à certaines bactéries aérobies et aux cellules des plantes et des animaux l’essentiel de leur énergie sous forme de molécules d’ATP. La notion de complexes impliqués dans ces processus émergea de la constatation que l’intégrité de la membrane n’est pas nécessaire au maintien d’un transport d’électrons couplé à une synthèse d’ATP. Des fragments de membrane, des préparations membranaires et vésiculaires (Electrons Transfer Particles, ETP) sont capables d’accomplir ces fonctions. Ces complexes sont au nombre de quatre et ils agissent successivement et de manière coordonnée (« coopérative »). Ils sont formés par l’assemblage d’environ 80 polypeptides, presque tous différents les uns des autres. La masse moléculaire de l’ensemble dépasse 2.000.000 de daltons. Les sous-unités protéiques des complexes I, III, IV (et de l’ATP synthase dont il sera question plus loin) sont codées par les génomes nucléaire et mitochondrial ; les sous-unités du complexe II, par le génome nucléaire. Au sein des complexes, une chûte de potentiel d’électrons à haute énergie est couplée à un pompage de protons de la matrice mitochondriale vers l’espace intermembranaire, raison pour laquelle ces machines moléculaires sont appellées « oxydo-réductases-pompes à protons ». Au nombre de quatre, elles sont numérotées de I à IV. Il existe un complexe V dont je parlerai plus loin. Avant de les examiner dans l’ordre, il faut rendre hommage au travail accompli par les chimistes et les physiciens qui ont établi les structures tridimensionnelles des complexes par diffraction des rayons X. Jusque dans les années 1980, cette technique n’avait été appliquée qu’à des protéines solubles (myoglobine, hémoglobine, lysozyme) pour lesquelles il est relativement aisé d’obtenir des cristaux. Le défi était tout autre avec des complexes membranaires essentiellement hydrophobes ; il fut résolu par la préparation de « cristaux bidimensionnels » réguliers, en s’inspirant des travaux effectués sur la rhodopsine, une protéine membranaire des cellules photo-réceptrices de la rétine (les « bâtonnets »). Pour établir la séquence en acides aminés des polypeptides constitutifs des complexes il fallut: (i) préparer des quantités substantielles de mitochondries de cœur de bœuf ; (ii) éliminer la membrane mitochondriale externe par traitement à la digitonine, et la matrice par traitement osmotique ; (iii) isoler la membrane interne par centrifugation ; (iv) solubiliser les complexes membranaires avec des détergents et des agents « chaotropiques » (qui dissocient les structures tertiaire et quaternaire des protéines) ; (v) séparer les polypeptides par chromatographie ; (vi) les séquencer. Il fallut ensuite isoler séparemment chacun des complexes et obtentir des structures suffisamment régulières et ordonnées. Dans les premiers diagrammes de diffraction à faible résolution (6,5 Angström), les détails des structures étaient peu visibles. L’utilisation de sources intenses de rayons X émanant des synchrotrons permit d’obtenir des diagrammes plus lisibles (résolution de 2,8 Angström). La première structure connue fut celle du centre réactionnel photosynthétique de la bactérie pourpre Rhodopseudomonas viridis.
Le point d’entrée dans la chaîne respiratoire des électrons fournis par le coenzyme NADH,H+ est le complexe I pompe à protons (NADH-CoQ réductase, NADH : ubiquinone oxydoréductase). C’est le plus volumineux des quatre complexes ; chez les mammifères, il est composé de 46 sous-unités – dont 14 sont codées par le génome mitochondrial – pour une masse d’environ 1 700 kDa. Sa structure par cristallographie aux rayons X fut établie à partir de différentes sources : mitochondries de cœur de bœuf, Escherichia coli (Ekaterina A. Baranova, Peter J. Holt, Leonid A. Sazanov, Institute of Science and Technology Austria, Klosterneuburg), Thermus thermophilus (Leonid A. Sazanov et Philip Hinchliffe, Medical Research Council, Dunn Human Nutrition Unit, Cambridge), la levure Yarrowia lipolytica (Volker Zickermann et coll. Institute of Biochemistry II, Medical School, Goethe Universität, Frankfurth). Les 2 e– fournis par le NADH sont transportés par une flavine adénine mononucléotide (FMN) dont le groupement prosthétique est la riboflavine (vitamine B2), un transporteur d’hydrogène (deux e– associés à deux H+) :
H2 ➔ 2H+ + 2e–
En acceptant la molécule d’hydrogène fournie par le NADH, le FMN passe successivement de la forme oxydée à la forme semiquinone (FMNH•) puis à la forme réduite (FMNH2). Les e– sont ensuite transférés à une série de protéines à Fe-S de types [2Fe-2S] et [4Fe-4S], puis à l’ubiquinone (coenzyme Q), un transporteur liposoluble à longue chaîne isoprénoïde, localisé à l’interface entre le complexe et la membrane mitochondriale interne. Un composé possédant les propriétés chimiques d’une quinone fut isolé en 1957 à partir de mitochondries de cœur de bœuf par Frederick L. Crane, un collaborateur de David E. Green (Institute for Enzyme Research, The University of Wisconsin) ; il le baptisa « coenzyme Q ». La même année, et indépendamment, Richard A. Morton (Johnston Chair of Biochemistry, University of Liverpool) caractérisa l’ubiquinone (quinone ubiquitaire) dans le foie de rats carencés en vitamines. L’ubiquinone est hydrophobe ; elle fixe deux e– et passe de la forme oxydée à la forme réduite ubiquinol (QH2) en acceptant deux protons issus de la matrice. Le complexe I-pompe à protons transfert 4 H+ de la matrice (mat) dans l’espace intermembranaire (inter).
NADH + Q10 + H+mat ➔ NAD+ + Q10H2
4 H+mat ➔ 4 H+inter
Références : Hinchliffe P, Sazanov LA Organization of iron-sulfur clusters in respiratory complex I (2005)
Sazanov LA, Hinchliffe P Structure of the Hydrophilic Domain of Respiratory Complex I from Thermus thermophilus (2006)
Baranova EA, Holt PJ, Sazanov LA Projection Structure of the Membrane Domain of Escherichia coli Respiratory Complex I at 8 Å Resolution (2007)
Zickermann V, Wirth C, Nasiri H, Siegmund K, Schwalbe H, Hunte C, Brandt U Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I (2015)
Crane FL, Hatefi Y, Lester RL, Widmer C Isolation of a quinone from beef heart mitochondria (1957)
Rappelons que la membrane mitochondriale interne est imperméable aux protons. Avant de quitter le complexe I, disons quelques mots des « protéines à fer-soufre » (protéines à Fe-S), découvertes par Helmut Beinert, un autre collaborateur de David Green. Elles possèdent au sein de leur structure deux, trois ou quatre atomes de fer liés à des anions sulfure (S2-) appartenant au groupe thiolate de la cystéine, et non à un groupe hème comme dans les cytochromes. En accord avec la variété de leurs structures, leurs potentiels redox sont variés. En 1964, John S. Rieske (Institute for Enzyme Research, University of Wisconsin, Madison) montra que les complexes bc1 et b6f de la chaîne respiratoire renferment une protéine à Fe-S (2Fe-2S) dont un atome de fer est lié à deux résidus histidines. Chaque atome de fer est dans un état d’oxydation qui lui est propre. Les protéines à Fe-S ont la capacité de transporter des électrons (un électron par transfert) et participent à des réactions d’oxydo-réduction dans les mitochondries et les chloroplastes chez les animaux, les plantes et les bactéries.
Référence : Rieske JS, MacLennan DH, Coleman R Isolation and properties of an iron-protein from the (reduced coenzyme Q)-cytochrome C reductase complex of the respiratory chain (1964)
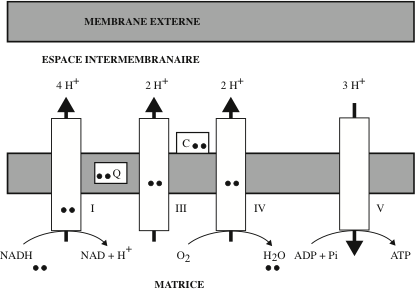 |
| Représentation simplifiée de la chaîne respiratoire. Les complexes I, III, et IV de la membrane interne des mitochondries sont des oxydoréductases–pompes à protons. Le complexe V est l’ATP synthase. Dans ce schéma, les électrons (deux points noirs) fournis par le NADH, c’est-à-dire par le cycle de Krebs et la β-oxydation des acides gras, libèrent leur énergie en suivant le trajet: complexe I – coenzyme Q (Q) – complexe III – cytochrome c (C) – complexe IV. Chaque saut d’énergie d’ampleur suffisante entraîne un pompage protons de la matrice vers l’espace intermembranaire par les oxydases–pompes à protons I, III, et IV. Les électrons fournis par une molécule de NADH permettent le pompage de dix protons. L’accepteur final des électrons dans le complexe IV est l’oxygène moléculaire. Le potentiel des protons stockés dans l’espace intermembranaire est consommé par le complexe V (ATP synthase) avec synthèse d’ATP. Dans cette figure, la stoechiométrie des réactions n’est pas prise en compte. |
Le complexe II (succinate coenzyme Q réductase) est constitué de quatre sous-unités : (i) la succinate déshydrogénase, le seul enzyme du cycle de Krebs à être localisé dans la membrane mitochondriale interne (les autres sont localisés dans la matrice) ; (ii) une flavoprotéine ; (iii) des protéines à Fe : S ; (iv) une ubiquinone réductase. Le groupe de Douglas C. Rees (Howard Hughes Medical Institute, California Institute of Technology() a déduit la structure du complexe II par cristallographie aux rayons X de la fumarate réductase d’Escherichia coli. Cet enzyme catalyse un transfert d’électrons vers le fumarate, l’accepteur final des électrons chez les organismes anaérobies. La structure cristallographique du complexe II de cœur de porc a été établie à une résolution de 2,4 angstrœm par Fei Sun et ses associés (Institute of Biophysics, Joint Research Group for Structural Biology, Tsinghua University and National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing).
succinate + FAD ➔ fumarate + FADH + H+
Le FADH2 délivre au coenzyme Q une paire d’e–, fournie par le succinate, et un H+, via trois centres à Fe-S :
FADH + H+ + Q10 ➔ FAD + Q10H2
Le complexe II est le second point d’entrée des électrons (fournis par le FADH2) dans la chaîne respiratoire. Le potentiel du couple redox FAD / FADH2 est de -0,20 V contre -0,32V pour le couple NAD+/NADH. Le complexe II ne contribue pas à la formation du gradient de concentration d’ions H+.
Références : Iverson TM, Luna-Chavez C, Cecchini G, Rees DC Structure of the Escherichia Coli Fumarate Reductase Respiratory Complex (1999)
Iverson TM, Luna-Chavez C, Schröder I, Cecchini G, Rees DC Analyzing your complexes: structure of the quinol fumarate reductase respiratory complex (1999)
Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z Crystal structure of mitochondrial respiratory membrane protein complex II (2005)
 |
|
Représentation simplifiée de la chaîne respiratoire. Les électrons fournis par le FADH2 (non représenté sur la Fig.) entrent dans la chaîne respiratoire au niveau du complexe II. Ils proviennent de la conversion de succinate (SUCC) en fumarate (FUM) par la succinate : ubiquinone oxydoréductase, le seul enzyme du cycle de Krebs localisé dans la membrane mitochondriale interne. Les électrons sont transférés successivement au coenzyme Q (Q), au complexe III, au cytochrome c (C) et au complexe IV. Les protons sont pompés dans l’espace intermembranaire par les complexes III et IV. |
Dans le complexe III (Coenzyme QH2-Cytochrome c réductase), comme dans le complexe IV, le transport des e– est assuré, non plus par des flavines comme dans les complexes I et II, mais par des cytochromes. Historiquement, les cytochromes furent les premiers transporteurs d’e– identifiés ; les cytochromes (a, a3 et c) Ils furent découverts par un médecin féru d’expérimentation, Charles A. MacMunn, en observant au spectroscope divers organismes et tissus (notamment des muscles). Les résultats de cet « amateur » furent soumis à une virulente critique de la part d’un grand « professionnel », Felix Hoppe-Seyler, fondateur et rédacteur en chef du Zeitschrift fûr Physiologische Chemie et pionnier de l’étude de l’hémoglobine et de la chlorophylle. C’est ainsi que la découverte des cytochromes tomba aux oubliettes pendant près de quarante ans. Lorsque David Keilin (Molteno Institute for Research in Parasitology, University of Cambridge) découvrit les cytochromes, il eut l’élégance de reconnaître l’antériorité de MacMunn. Ce dernier avait découvert les cytochromes a, a3 et c ; dans les années 40 et 50, Ichiro Y. Sekuzu et Kazuo Okuniki (Department of Biology, University of Osaka) découvrirent les cytochromes c1 et b. Le groupe hème de ces hémoprotéines, constitué d’une porphyrine et d’un atome de fer ionisé, leur confère la capacité d’accepter ou de donner un e– avec passage réversible de l’état ferreux à l’état ferrique.
Références : MacMunn CA VI. Researches on myohamatin and the histohaematins (1886)
Keilin D. On cytochrome, a respiratory pigment, common to animals, yeast and higher plants (1925)
Keilin D A comparative study of turacin and haematin and its bearing on cytochrome (1926)
Keilin D Le cytochrome pigment respiratoire intracellulaire commun aux micro-organismes, aux plantes et aux animaux (1927)
Sekuzu I, Takemori S, Orii Y, Okunuki K Studies on cytochrome a: IV. Reaction of cytochrome a with cytochromes c and c1 (1960)
Takemori S, Sekuzu I, Okunuki K Studies on cytochrome a VII. Physico-chemical properties of purified cytochrome a (1961)
La structure cristallographique de la CoQH2-Cytochrome c réductase a été élucidée chez la levure Saccharomyces cerevisiae et dans les mitochondries de cœur de bœuf par les groupes de Hartmut Michel (Molecular Membrane Biology Department, Max Planck Institute for Biophysics, Frankfurt am Main, prix Nobel de chimie 1988) et de Johann Deisenhofer (Department of Biophysics, The University of Texas Southwestern Medical Center, Dallas, prix Nobel de chimie 1988). Chez les mammifères, c’est un volumineux dimère dont les monomères comportent 11 sous-unités différentes. Les cofacteurs (deux cytochromes b, un cytochrome c1, une protéine de Rieske à 2Fe-2S) sont associés à 3 sous-unités codées par le génome mitochondrial.
Références : Hunte C, Koepke J, Lange C, Rossmanith T, Michel H Structure at 2.3 A resolution of the cytochrome bc(1) complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment (2000)
Lange C, Hunte C Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c (2002)
Zara V, De Blasi G, Ferramosca A Assembly of the Multi-Subunit Cytochrome bc1 Complex in the Yeast Saccharomyces cerevisiae (2022)
Xia D, Yu CA, Kim H, Xia JZ, Kachurin AM, Zhang L, Yu L, Deisenhofer J Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria (1997)
Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex (1998)
Solmaz SR, Hunte C Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer (2008)
Marten Wikström (Department of Medical Chemistry, University of Helsinki) et Gerald T. Babcock (Department of Chemistry, Michigan State University, East Lansing) ont élucidé le trajet des e– au sein du complexe III, du CoQH2 au cytochrome c. Dans une revue publiée en 1990, Bernard L. Trumpower (Geisel School of Medicine, Dartmouth) a décrit dans le détail le rôle joué par la navette Q/QH2 dans le transfert d’électrons, des complexes I et II vers le complexe III. Présent chez une variété d’organismes, le « protonmotive Q cycle » est le plus important mécanisme de transduction d’énergie. J’ai rappelé, dans un paragraphe précédent, les circonstances de la découverte de l’ubiquinone (1,4 benzoquinone), connue aussi sous l’appellation de coenzyme Q, ou CoQ10, par référence au dix unités isoprène de la chaîne latérale, qui ancre le coenzyme dans la bicouche phospholipidique. Le CoQH2 porte 2H+ et 2e– ; au sein du complexe III, il libère 2 H+ dans l’espace inter-membranaire et fournit un e– au cytochrome c par l’intermédiaire d’une protéine à Fe :S ; il fournit un autre e– au cytochrome b et charge 2H+ venant de la matrice. Le cytochrome c est une protéine soluble de 12.500 daltons dont le cofacteur est un hème de type c ; il est localisé dans l’espace inter-membranaire et transfert un e– vers le complexe IV et l’accepteur final, l’O2. Au total, le complexe III injecte quatre H+ dans l’espace inter-membranaire et réduit deux molécules de cytochrome c.
Q10H2 + 2 Ccox + 2 H+mat ➔ Q10 + 2 Ccred + 4 H+inter
Références : Wikström M, Babcock GT Cell Respiration. Catalytic Intermediates (1990)
Trumpower BL The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex (1990)
Crane F L, Sun IL, Sun EE The essential functions of coenzyme Q (1993)
Ding H, Daldal F, DuttonPL Ion pair formation between basic residues at 144 of the Cyt b polypeptide and the ubiquinones at the Qo site of the Cyt bc1 complex (1995)
Turunen M, Olsson J, Dallner G Metabolism and function of coenzyme Q (2004)
Darrouzet E, Moser CC, Dutton PL, Daldal F Large scale domain movement in cytochrome bc(1): a new device for electron transfer in proteins (2001)
Le complexe IV (Cytochrome c Oxydase) appartient à la superfamille des oxydases à hème-cuivre. C’est par lui que tout a commencé ! Je parle de la « saga » des travaux sur la respiration cellulaire et la production d’énergie, qui se sont étalées sur des décennies. Le complexe IV est l’Atmungsferment, découvert en 1925 par Otto H. Warburg (prix Nobel de physiologie ou médecine 1931), rebaptisé « cytochrome oxydase » en 1929, partiellement purifié à partir de cœur de bœuf, en 1941, par von Eijiro Yakushiji et Kazuo Okunuki (Iwata Institut fûr Pflanzenbiochemie). Sa structure à l’échelle atomique fut établie par Tomitake Tsukihara et coll. (Institute for Protein Research, Osaka University) ; le complexe IV comporte 13 sous-unités polypeptidiques, dont trois pour la partie catalytique. Le groupe de Hartmut Michel (Molecular Membrane Biology Department, Max Planck Institute for Biophysics, Frankfurt am Main, prix Nobel de chimie 1988) a établi la structure tridimensionnelle du complexe IV (trois sous-unités) de Paracoccus denitrificans. Le complexe IV reçoit quatre e–, transférés depuis le complexe III par le transporteur soluble cytochrome c ; quatre molécules de cytochrome c délivrent, successivement, un e– à une paire d’ions cuivre (Cu2+) puis à l’hème du cytochrome a et enfin au centre catalytique : ions Cu2+ et cytochrome a3. Les quatre e– sont transférés à l’accepteur final O2 ; la chute de potentiel redox entre le cytochrome c et l’O2 est couplée au pompage dans l’espace inter-membranaire de quatre H+ ; quatre autres H+ sont consommés pour former deux molécules de H2O) :
4 Ccred + 8 H+mat + O2 ➔ 4 Ccox + 2 H2O + 4 H+inter
Références : Warburg OH Über Eisen, den sauerstoff-übertragenden Bestandteil des Atmungsferments (1925)
Yakushiji E, Okunuki K Isolierung der a-Komponente des Cytochroms und ihre Eigenschaften (1941)
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 Å (1995)
Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S The Whole Structure of the 13-Subunit Oxidized Cytochrome c Oxidase at 2.8 A (1996)
Iwata S, Ostermeier C, Ludwig B, Michel H Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans (1995)
L’analyse chimique de préparations purifiées a révélé que le rapport protéines/phospholipides de la membrane mitochondriale interne est élevé – le plus élevé des membranes cellulaires – ce qui laissait entrevoir des contacts étroits entre complexes respiratoires individuels. En 1955, Britton Chance et Graham R. Williams (Johnson Foundation for Research in Medical Physics, School of Medicine, University of Pennsylvania) émirent l’hypothèse que la chaîne respiratoire est formée de super-complexes. Bernard L Trumpower (Department of Biochemistry, Darmouth Medical School, Hanover) fut l’un des premiers à caractériser un super-complexe chez Paracoccus denitrificans.
Références : Chance B, Williams GR A Method for the Localization of Sites for Oxidative Phosphorylation (1955)
Berry EA, Trumpower BL Isolation of ubiquinol oxidase from Paracoccus denitrificans and resolution into cytochrome bc1 and cytochrome c-aa3 complexes (1985)
Sone N, Sekimachi M, Kutoh E Identification and properties of a quinol oxidase super-complex composed of a bc1 complex and cytochrome oxidase in the thermophilic bacterium PS3 (1987)
Les progrès techniques dans la séparation et la visualisation des ensembles supramoléculaires ont fait progresser nos connaissances dans le domaine des supercomplexes mitochondriaux. Pour pouvoir les étudier, il faut : (i) préserver les fragiles interactions protéines-protéines ; (ii) éviter les dommages infligés aux structures subcellulaires par les faisceaux d’électrons à haute intensité utilisés en diffraction aux rayons X. La première difficulté a été résolue par l’emploi de l’électrophorèse en gel de polyacrylamide dite « Blue Native ». Alexanne Cuillerier et Yan Burelle (Faculty of Health science, University of Ottawa) ont mis au point une technique combinant les avantages des « Clear Native » et « Blue Native » Polyacrylamide Gel Electrophoresis. Les supercomplexes de la membrane mitochondriale interne sont solubilisés avec un détergent non ionique (digitonine) ; ils sont colorés en bleu par un bref contact avec le colorant anionique Bleu de Comassie (Comassie Brillant Blue). L’électrophorèse est conduite en conditions non dénaturantes (en l’absence de Sodium dodécyl sulfate). La séparation peut être complétée par une électrophorèse bidimensionnelle. Runyu Guo et coll. (Advanced Innovation Center for Structural Biology, School of Life Sciences, Tsinghua University, Beijing) ont utilisé la centrifugation en gradient de densité pour isoler un supercomplexe de mitochondries de cellules humaines en culture.
Références : Jha P, Wang X, Auwerx J Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) (2016)
Cuillerier A, Burelle Y Hybrid Clear/Blue Native Electrophoresis for the Separation and Analysis of Mitochondrial Respiratory Chain Supercomplexes (2019)
Guo R, Zong S, Wu M, Gu J, Yang M Architecture of Human Mitochondrial Respiratory Megacomplex I(2)III(2)IV(2) (2017)
La seconde difficulté a été résolue par l’usage de la cryomicroscopie électronique (avec une résolution inférieure à 4 Å). Les structures supramoléculaires (respirasomes) ont été étudiées chez les mammifères (homme, bœuf, rat), dans divers organes (cerveau, foie, rein, muscle, cœur, fibroblastes), chez les végétaux, les champignons, les levures, les bactéries et le nématode Caenorhabditis elegans. Pour répondre dans les plus brefs délais aux besoins énergétiques de la cellule, la chaîne respiratoire mitochondriale adopte l’organisation des complexes I à IV en supercomplexes, ce qui limiterait les phénomènes de diffusion. La cohésion au sein de ces structures supramoléculaires est assurée par les interactions fortes entre protéines, et par la présence d’un phospholipide particulier, la cardiolipine. Découverte en 1941 dans le muscle cardiaque par Mary C. Pangborn (Division of Laboratories and Research, New York State Department of Health, Albany), la cardiolipine (glycérol bisphosphatidyl) représente près de 20% des phospholipides de la membrane mitochondriale interne, dont elle assure l’imperméabilité aux protons.
Références : Iwasaki T, Matsuura K, Oshima T Resolution of the aerobic respiratory system of the thermoacidophilic archeon, Sulfolobus sp. Strain 7. I. The archeal terminal oxidase supercomplex is a functional fusion of respiratory complexes III and IV with no c-type cytochrome (1995)
ruel C, Brasseur R, Trumpower BL Subunit 8 of the Saccharomyces cerevisiae cytochrome bc1 complex interacts with succinate-ubiquinone reductase complex (1996)
Boumans H, Grivell LA, Berden JA The respiratory chain in yeast behaves as a single functional unit (1998)
Schagger H, Pfeiffer K Supercomplexes in the respiratory chains of yeast and mammalian mitochondria (2000)
Eubel H, Heinemeyer J, Sunderhaus S, Braun HP Respiratory chain supercomplexes in plant mitochondria (2004)
Reifschneider NH, Goto S, Nakamoto H, Takahashi R, Sugawa M, Dencher NA, Krause F Defining the Mitochondrial Proteomes from Five Rat Organs in a Physiologically Significant Context Using 2D Blue-Native/SDS-PAGE (2006)
Krause F OXPHOS Supercomplexes: Respiration and Life-Span Control in the Aging Model Podospora anserine (2006)
Schäfer E, Dencher NA, Vonck J, Parcej DN Three-dimensional structure of the respiratory chain supercomplex I1II2IV1 from bovine heart mitochondria (2007)
Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA Respiratory active mitochondrial supercomplexes (2008)
Lombardi A, Silvestri E, Cioffi F, Senese R, Lanni A, Goglia F, de Lange P, Moreno M Defining the transcriptomic and proteomic profiles of rat ageing skeletal muscle by the use of a cDNA array, 2D- and Blue native-PAGE approach (2009)
Sunderhaus S, Klodmann J, Lenz C, Braun HP Supramolecular structure of the OXPHOS system in highly thermogenic tissue of Arum maculatum (2010)
Althoff T, Mills DJ, Popot JL, Kühlbrandt W Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1II2IV1 (2011)
Schäfer E, Seelert H, Reifschneider NH, Krause F, Dencher NA, Vonck J Architecture of active mammalian respiratory chain supercomplexes (2017)
Pangborn M Isolation and purification of a serologically active phospholipid from beef heart (1942)
Ci-dessus, j’ai énuméré les complexes dans l’ordre dans lequel ils interviennent entre le point d’entrée dans la chaîne respiratoire (NADH) et le point de sortie (l’oxygène apporté par la respiration pulmonaire). Pour déterminer cet ordre, on a utilisé deux approches expérimentales: (i) la mesure des potentiels redox des groupes prosthétiques ; le résultat a permis le classement des complexes par ordre de potentiel décroissant; (ii) l’utilisation d’agents découplants (antimicyne A, roténone, cyanure, amytal) qui permet de dissocier le transfert d’électrons de la synthèse d’ATP; ils interrompent le transfert des électrons en un point défini de la chaîne respiratoire (l’antimicyne A, par exemple, interrompt le flux d’électrons entre le NADH et l’ubiquinone). Le grand spécialiste de la mesure des proportions de forme oxydée et de forme réduite des transporteurs était le physico-chimiste Britton Chance (E. R. Johnson Foundation, The University of Pennsylvania). Dans les années 1960, il perfectionna les techniques d’analyse non invasives de matériel biologique (résonance magnétique nucléaire, spectroscopie et fluorométrie) et mit au point d’élégantes techniques spectroscopiques de mesure des formes oxydées et réduites. L’addition d’inhibiteurs modifiait ces proportions. Accessoirement, Chance, qui était un passionné de navigation à voile, remporta une médaille d’or aux Jeux olympiques d’Helsinki, en 1952.
La liaison fonctionnelle entre complexes est assurée par des transporteurs d’électrons mobiles : le coenzyme Q entre les complexes I et III ; le cytochrome c, dans l’espace intermembranaire, entre les complexe III et IV. Le passage des électrons d’un complexe au suivant s’accompagne d’une diminution graduelle de l’énergie libre stockée, ce qui correspond à une augmentation du potentiel redox E°. Si la diminution de E° dépasse le seuil de 300 millivolts, l’énergie libérée permet le pompage de protons de la matrice vers l’espace intermembranaire (les complexes I, III et IV sont des oxydo réductases-pompes à protons).
Bilan
Etablissons le bilan énergétique de la chaîne respiratoire. Au point d’entrée de la chaîne respiratoire, le complexe I catalyse la réaction redox :
NADH + H+ ➔ NAD+ + 2 H+ + 2 e– (E0 = -0,32V)
A la sortie de la chaîne respiratoire, le complexe IV catalyse la réaction redox :
1/2 O2 + 2 H+ + 2 e– ➔ H2O (E0 = +0,82V)
soit, en additionant les deux réactions :
NADH + H+ + 1/2 O2 ➔ NAD+ + H2O
avec une différence de potentiel de réduction standard :
ΔE0 = +1,14V
ce qui correspond à une variation d’énergie libre standard :
ΔG0 = -220kJ.mol-1
Cette quantité d’énergie est théoriquement suffisante pour synthétiser 7 molécules d’ATP (par l’ATP synthase). Chaque fois qu’au sein d’une oxydase-pompe à protons le saut d’énergie électrique est supérieur ou égal à 300 millivolts, des protons sont transférés de la matrice vers l’espace intermembranaire. C’est ainsi que pour chaque molécule de NADH entrant dans la chaîne respiratoire, dix protons sont pompés, créant un potentiel de membrane.
Synthétiser de l’ATP
La découverte de la relation entre la chaîne respiratoire et les oxydations phosphorylantes date de la fin des années 1940. Dans les mitochondries, les chloroplastes et chez certaines bactéries, l’énergie d’électrons extraits des nutriments est utilisée pour synthétiser de l’ATP. Lorsque la chaîne respiratoire est alimentée en électrons, de l’ATP est synthétisé. Au cœur de ce processus, il y a un complexe membranaire, l’ATP synthase, une « Splendid Molecular Machine », comme l’appelait Paul D. Boyer. Il fallut plus d’un demi siècle pour comprendre comment fonctionne ce système de conversion d’énergie osmotique en énergie chimique, comment l’ATP synthase utilise la force proton-motrice pour faire de l’ATP. La synthèse d’ATP à partir d’ADP et de phosphate inorganique (Pi ou P)
ADP + P ➔ ATP + H2O
est une réaction thermodynamiquement défavorable. On dit qu’elle est endergonique ; la variation d’énergie libre ΔG > 0 est positive. Elle ne devient possible que si elle est couplée à une réaction thermodynamiquement favorable :
X~P + H2O ➔ X + P
La liaison riche en énergie d’hydrolyse est symbolisée par le signe ~. La réaction ci-dessus est exergonique ; sa variation d’énergie libre est négative : ΔG < 0. Lorsque les deux réactions sont couplées, la molécule d’eau n’apparait plus dans l’échange :
X~P + ADP ➔ X + ATP
Si la variation d’énergie libre ΔG des deux réactions est négative et d’ampleur suffisante, de l’ATP est synthétisé. Les composés X~P du métabolisme intermédiaire sont le plus souvent les produits d’estérification d’un thiol (R-SH) et d’un acide carboxylique (R-COOH). Le composé résultant posséde une liaison thioester (-CO-S-) « riche en énergie » dont l’hydrolyse libère 14 kcal par molécule-gramme, la même quantité d’énergie que l’hydrolyse de la liaison pyrophosphate terminale de l’ATP. Un « intermédiaire riche en énergie » fut identifié par Efraïm Racker (New York University Medical School) dans la réaction de conversion d’un dialdéhyde, le glyoxal ou éthanedial : CHO-CHO en acide glycolique (acide hydroxy acétique) : CHOH-COOH, catalysée par la glyoxalase. Il s’agissait d’un dérivé carboxylé du pseudo-tripeptide glutathion (glutamyl-cystéinyl-glycine) possèdant une liaison thioester :
R-SH + COOH-CHOH ➔ R-S-COO-CHOH
Dans la réaction ci-dessus (non équilibrée) R-SH est le glutathion. Des dérivés de thiols et d’acides carboxyliques interviennent dans d’autres voies métaboliques comme la glycolyse, le cycle de l’acide citrique, le cycle du glyoxylate…
En 1897, Hans E.A. Buchner proposa d’ajouter du saccharose à des broyats de levure, préparés par le chimiste Martin Hahn, pour améliorer leur conservation. Eduard Buchner, le frère de Hans, observa que l’addition de sucre provoquait une fermentation qu’il attribua à la présence d’un « ferment actif », baptisé « zymase ». C’était le premier jalon dans la découverte de la glycolyse anaérobie, la première voie métabolique à être élucidée. Au cours de ce processus, présent chez les bactéries, les mycètes, les végétaux et les animaux, du sucre est transformé en alcool ou en acide lactique et de l’ATP est synthétisé sans intervention (apparente) d’oxygène. Les travaux d’Eduard Buchner (prix Nobel de chimie en 1907), de Carl F. et Gerty T. Cori, de Gustav G. Embden, d’Otto H. Meyerhof, de Jacob K. Parnas, de Carl Neuberg et d’Otto Warburg aboutirent à l’élucidation des dix étapes de ce processus fondamental chez tous les êtres vivants. Le produit final de la glycolyse varie selon les conditions prévalant dans le cytosol des cellules eucaryotes : en aérobiose, le produit final est une molécule de pyruvate (en réalité deux par molécule de glucose) qui entre dans le cycle de Krebs sous forme d’acétyl-CoA ; en anaérobiose, le pyruvate est transformé en lactate (fermentation lactique dans les muscles), ou en alcool (fermentation alcoolique chez les levures).
C’est en étudiant le mode de récupération de l’énergie dans la glycolyse que Paul D. Boyer (Molecular Biology Institute, University of California, Los Angeles) et Efraïm Racker et Isidore Krimsky mirent en évidence, en 1952, l’intervention d’un intermédiaire enzymatique acylé. Au cours de l’étape VI, le groupe aldéhyde du glycéraldéhyde 3-phosphate est oxydé en groupe carboxylate du 3-phosphoglycérate, catalysée par la glycéraldéhyde-3-phosphate déshydrogénase, la seule oxydo-réductase (ou transférase d’électrons) de la voie métabolique. La réaction se déroule en deux étapes au cours desquelles deux intermédiaires réactionnels covalents interviennent : (i) un premier intermédiaire se forme lorsque le groupe sulfidryl d’un résidu cystéine du site catalytique de de la glycéraldéhyde 3-phosphate déshydrogénase est acylé par le groupe carbonyl du glycéraldéhyde 3-phosphate (catalyse covalente) ; cet intermédiaire enzymatique acylé est attaqué par une molécule de phosphate inorganique avec formation de 1,3-bis-phosphoglycérate. C’était la première fois que l’on mettait en évidence le rôle joué par une liaison thiol-ester. (ii) un second intermédiaire phosphorylé se forme lors du tranfert d’un groupe phosphoryl sur l’ADP. La synthèse d’ATP est rendue possible grâce à l’énergie libérée par l’hydrolyse de la liaison anhydride mixte d’acide phosphorique et d’acide carboxylique sur le carbone C1 du 1,3-bis-phosphoglycérate. La différence de potentiel entre le couple redox glycéraldéhyde phosphate/acide phosphoglycérique et le couple redox :
NAD+ + 2H+ + 2e– ↔ NADH + H+
supérieure à 300 millivolts, permet la synthèse électrochimique d’une molécule d’ATP (deux par molécule de glucose). En 1953, Edward C. Slater (Department van Biochemie, Universiteit van Amsterdam), Boyer et d’autres biochimistes se tournèrent vers un mécanisme similaire mettant en jeu deux intermédiaires covalents pour la synthèse d’ATP dans les mitochondries. L’hypothèse du couplage chimique avec un intermédiaire riche en énergie, ou hypothèse « chimio-chimique » orienta les recherches des biochimistes pendant deux décades.
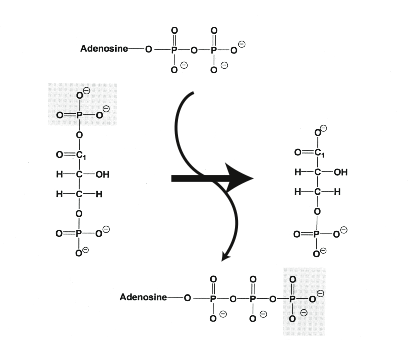 |
| Phosphorylation au niveau du substrat dans la glycolyse. La glycéraldéhyde 3-phosphate déshydrogénase (G3PDH) catalyse l’oxydation du glycéraldéhyde 3-phosphate (G3P) en 1,3-bis-phosphoglycérate (molécule de gauche). La phosphoglycérate kinase catalyse le transfert du groupe phosphoryl du carbone C1 sur une molécule d’ADP, avec récupération de l’énergie de la liaison anhydride mixte d’acide, et formation d’une molécule d’ATP et de 3-phosphoglycérate (molécule de droite). Les deux réactions sont couplées. |
Dans le laboratoire de Carl et Gerty Cori (Washington University School of Medicine), Mildred Cohn, pionnière de l’emploi des radioisotopes en biologie, montra en 1963 qu’en présence de mitochondries s’opère un échange rapide d’oxygène (18O) entre le phosphate inorganique et l’eau. Boyer s’inspira d’un protocole expérimental similaire avec 18O et 32P et montra que l’ensemble des oxydations phosphorylantes était réversible. Il attribua l’échange rapide d’oxygène à l’hydrolyse d’un intermédiaire covalent riche en énergie par du phosphate inorganique. Le domaine F1 de l’ATP synthase catalyse la réaction réversible :
ADP + Pi ↔ ATP + H2O
comme le montre le calcul des constantes d’équilibre des vitesses de synthèse et d’hydrolyse. Lorsque la réaction ci-dessus fonctionne dans le sens de l’hydrolyse d’ATP en présence d’oxygène, le Pi peut incorporer jusqu’à quatre atome d’18O. En faisant abstraction des nucléotides, dans la réaction :
Pi ↔ HOH
l’échange d’18O refléte quantitativement la vitesse de formation et d’hydrolyse de l’ATP. Avec le complexe F1, l’échange se déroule sans apport d’énergie. Avec des particules submitochondriales (complexe FoF1), l’échange se déroule en présence d’agents découplants. L’incorporation de plusieurs atomes d’18O dans Pi révèle que la liaison γ−pyrophosphate a été hydrolysée et reformée plusieurs fois. D’autre part, la vitesse d’échange d’18O est plus élevée que la vitesse de réversibilité de la réaction, ce qui est compatible avec l’intervention d’un intermédiaire covalent dont le clivage par une molécule de Pi donnerait de l’ATP. Il fallut plus d’une décennie pour comprendre que cette interprétation n’était pas la bonne et que l’échange d’oxygène était dû au relargage d’une molécule d’ATP fixée sur un site catalytique de l’ATP synthase.
Dans les années 1960, Boyer poursuivi sa quête infructueuse en caractérisant une protéine mitochondriale fixant du 32P sur un résidu histidine et servant d’intermédiaire entre le phosphate inorganique et l’ATP. Il ne s’agissait pas de l’intermédiaire des oxydations phosphorylantes recherché mais d’un intermédiaire de la succinyl CoA synthetase qui, dans le cycle de Krebs, synthétise de l’ATP (ou du GTP) à partir d’un nucléotide diphosphate et de Pi.
Hypothèse chimio-osmotique
« Max Planck (…) remarked that a new scientific idea does not triumph by convincing its opponents, but rather because its opponents eventually die. »
Peter Mitchell
Le ton vengeur de la phrase de Peter Mitchell citée en exergue est une conséquence de l’opposition qu’il dut affronter lorsqu’il présenta son hypothèse chimio-osmotique. En biologie, de grandes découvertes qui ont radicalement changé le cours de nos connaissances ont souvent fait l’objet de publications de taille modeste. En 1953, la description de la structure en double hélice de l’ADN par James Watson et Francis Crick fit l’objet d’un article de deux pages dans la revue Nature. Ce fut également le cas pour l’article de Peter D. Mitchell, dans lequel il critiquait l’hypothèse chimio-chimique en faisant remarquer que les enzymes de la glycolyse anaérobie sont des protéines cytosoliques, donc solubles alors que les complexes de la voie respiratoire dépendent pour leur fonctionnement de l’intégrité de la membrane ; toute altération mécanique ou chimique (détergents) de la membrane mitochondriale interne supprime le transport des électrons et la synthèse d’ATP. Mitchell mettait en doute l’intervention d’un intermédiaire chimique. Dépassant le stade de la critique, il proposa une élégante hypothèse mécanistique. L’énergie nécessaire à la synthèse d’ATP proviendrait d’une force proton-motrice créée par un gradient de concentration en protons de part et d’autre de la membrane interne ; un mécanisme analogue à la chimio-osmose (diffusion de molécules d’eau à travers une membrane) utiliserait l’énergie cinétique du passage de protons à travers la membrane ; ce passage se ferait à travers le complexe de l’ATP synthase et l’énergie libérée par le flux des protons vers la matrice mitochondriale serait couplée à la synthèse d’ATP. L’hypothèse chimio-osmotique provoqua un scepticisme général dans la communauté des « mitochondriaques ». Ceux-ci considéraient avec suspicion tout ce qui venait de Mitchell, ce franc-tireur de la biochimie qui finançait lui-même ses recherches (avec l’argent de sa femme) sans recourir à l’argent public. Avec sa collaboratrice Jennifer Moyle, ce scientifique hors normes avait fondé une structure de recherche privée : les Glynn Research Laboratories. Au fil du temps, des résultats expérimentaux étayant l’hypothèse chimio-osmotique furent publiés. Mitchell et Moyle démontrèrent une corrélation entre le déplacement des électrons le long de la chaîne de respiratoire et le pompage de protons de la matrice vers l’espace intermembranaire.
Références: Mitchell PD Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism (1961)
Mitchell PD, Moyle J Stoichiometry of proton translocation through the respiratory chain and adenosine triphosphatase systems of rat liver mitochondria (1965)
L’appellation « adénosine triphosphatase » dans le titre de l’article de Mitchell et Moyle rappelle que le fonctionnement du complexe est réversible ; il fonctionne comme une ATP synthase lorsque les protons passent de l’espace intermembranaire vers la matrice, et comme une ATPase lorsque les protons suivent le chemin inverse.
Alors qu’il séjournait dans le laboratoire d’Efraïm Racker (Biochemistry Department, Cornell University), André T. Jagendorf (College of Agriculture and Life Sciences, Cornell University) prit connaissance de l’hypothèse de Mitchell. Il la transposa au cas de la photophosphorylation avec synthèse d’ATP dans les chloroplastes (voir le chapitre « Chloroplastes »). En 1966, Jagendorf et Ernest G. Uribe purifièrent des thylakoïdes de feuilles d’épinard et les incubèrent à l’obscurité dans une solution de pH acide ; les protons franchirent la membrane par diffusion passive et s’accumulèrent dans les disques jusqu’à l’équilibre avec le milieu. Lorsque les thylakoïdes chargés en protons furent incubés dans l’obscurité, dans une solution de pH plus élevé, en présence d’ADP et de 32Pi, Jagendorf et Uribe observèrent une accumulation d’AT32P dans le stroma ; le relâchement des protons s’accompagnait d’une synthèse d’ATP. Walther Stoeckenius (University of California, San Francisco) et Ephraïm Racker s’associèrent pour préparer des liposomes dans la membrane desquels ils insérèrent de la bactériorhodopsine et la F1Fo-ATPase. Racker et ses collaborateurs maîtrisaient les techniques de purification de l’ATPase de cœur de bœuf et de préparation des liposomes. Le succès de l’expérience reposait sur le bon vouloir des deux protéines à adopter l’orientation correcte au moment de leur insertion in vitro dans la membrane liposomiale. Et le miracle se produisit ! Après éclairement, des protons furent pompés dans les liposomes ; en présence d’ADP et de Pi dans le milieu, de l’ATP fut synthétisé !
Références: Jagendorf AT, Uribe EG ATP formation caused by acid-base transition of spinach chloroplasts (1966)
Racker E, Stoeckenius W Reconstitution of purple membrane vesicle catalyzing light-driven proton uptake and adenosine triphosphate formation (1974)
En 1977, le modèle chimio-osmotique de Peter Mitchell était à peu près unanimement accepté. L’énergie libérée par le transport d’électrons dans la chaîne respiratoire mitochondriale, dans les photosystèmes chloroplastiques ou par la bactériorhodopsine membranaire est utilisée pour pomper des protons à travers une membrane (imperméable aux protons) avec génération d’un potentiel électrostatique. L’énergie générée par le retour des protons à travers le complexe membranaire de l’ATP synthase produit de l’ATP. Le vainqueur de la « Great Chemi-osmotic War » (selon l’expression d’un concurent malheureux) reçut le prix Nobel de Chimie en 1978. Restait à élucider le mécanisme de fonctionnement de l’ATP synthase. Mitchell proposa une interaction directe entre le flux de protons et le site catalytique de l’enzyme ; la réalité allait se révéler bien plus sophistiquée.
Avec le concept de la catalyse rotationnelle, Paul D. Boyer proposa un modèle très différent de celui de Mitchell. L’énergie délivrée par le flux de protons provoquerait un changement de conformation du site catalytique de l’ATP synthase, détachant ainsi une molécule d’ATP fermement liée à ce site. Cette hypothèse lui fut inspirée par le modèle de la myosine. Les mouvements cellulaires sont rendus possible par l’interaction de deux protéines filamenteuses : l’actine et la myosine. La myosine est un complexe protéique abondant dans le cytoplasme des cellules contractiles des muscles lisses et des muscles striés. Elle est formée de deux chaînes lourdes (2.000 résidus d’acides aminés) comprenant, de l’extrémité carboxy-terminale à l’extrémité amino-terminale, la queue – des hélices α surenroulées, sauf à leurs extrémités carboxy-terminales -, le cou – deux chaînes légères associées à chacune des deux chaînes lourdes -, et la tête – deux domaines globulaires. L’ensemble a la forme d’un bâtonnet de 180 nanomètres de long. Le domaine globulaire est un moteur moléculaire possédant un « site nucléotides » (ATP ou ADP + Pi) et un « site actine ». L’actine globulaire (actine G) est associée à un cation divalent (Ca2+ ou Mg2+) et à une molécule d’ATP dont l’hydrolyse déclenche l’association des molécules d’actine G en ploymère hélicoïdal d’actine filamenteuse (actine F). La progression de la myosine le long des filaments d’actine se fait par l’intermédiaire de ses domaines globulaires. Les têtes de myosine ayant fixé une molécule d’ADP + Pi sont en « configuration haute énergie », liées au filament d’actine. Après relarge d’ADP + Pi la tête revient en « configuration basse énergie » et le filament d’actine coulisse par rapport à celui de myosine. La fixation d’une molécule d’ATP par les domaines globulaires de la myosine entraîne un changement conformationnel qui les détache du filament d’actine. Les cinétiques de la réaction d’échange
Pi ↔ HOH
dans l’ATPase des têtes de la myosine et l’ATP synthase présentent des analogies. Boyer en déduisit que l’ATP synthase devait avoir un « site nucléotide » analogue à celui de la myosine. Il proposa un concept catalytique dans lequel l’énergie délivrée par le flux de protons provoquerait un changement de conformation du site catalytique de l’ATP synthase, ce qui aurait pour effet de détacher une molécule d’ATP fermement liée au site nucléotide.
L’hypothèse de la catalyse conformationnelle, publiée en 1973 par Paul Boyer, Richard L. Cross et William Momsen, rencontra d’emblée une certaine opposition ; elle fut réfutée par Peter Mitchell et l’article fut refusé par le Journal of Biological Chemistry ! Boyer étant membre de la National Academy of Sciences, il réussit à le faire accepter par les Proceedings de cette société sous le titre : « A New Concept for Energy Coupling in Oxidative Phosphorylation Based on a Molecular Explanation of the Oxygen Exchange ». Tenant compte des critiques formulées par Mitchell, Boyer publia en 1975 une version plus élaborée de son hypothèse. Je n’entrerai pas dans le détail des expériences de cinétique enzymatique réalisées par les collaborateurs de Boyer. Ce qui finit par imposer son hypothèse, c’est qu’un certain nombre d’anomalies étaient difficilement explicables dans le cadre de la cinétique classique des enzymes solubles. Par exemple : (i) en absence d’ADP dans le milieu d’incubation, l’ATP ne se détachait pas ; (ii) en absence d’ATP dans le milieu d’incubation, la réaction :
ADP + P ➔ ATP + H2O
s’arrêtait. Celik Kayalar, dans le laboratoire de Paul Boyer, proposa d’expliquer l’une de ces anomalies en faisant appel au concept de coopérativité, c’est-à-dire en supposant que le comportement du site ATP était influencé par un site ADP + Pi voisin. Le concept de régulation allostérique de l’activité enzymatique, proposé au milieu des années 1960 par Jacques Monod, Jean-Pierre Changeux et Jeffries Wyman (Institut Pasteur, Paris), met en jeu des protéines oligomériques formées de protomères symmétriquement semblables. Chaque protomère posséde un site catalytique fixant le substrat, un site allostérique fixant un agoniste et un cycle inhibiteur ; la fixation de l’agoniste sur le site allostérique provoque un changement de conformation du protomère dont le site actif peut fixer le substrat. Nous verrons plus loin que le changement de conformation subi par le site catalytique de l’ATP synthase ne répond pas exactement à ce modèle. En 1977, Celik Kayalar et Jan Rosing proposèrent un modèle pour l’ATP synthase de particules sub-mitochondriales à (au moins) deux sites catalytiques agissant de manière séquentielle et coopérative. En 1981, Boyer présenta le postulat de la catalyse conformationnelle aux Gordon Research Conferences. Le modèle à trois sites catalytiques fut proposé l’année suivante par Paul Boyer, Michael J. Gresser et Jill A. Myers dans un article intitulé « Catalytic site cooperativity of beef heart mitochondrial F1 adenosine triphosphatase. Correlations of initial velocity, bound intermediate, and oxygen exchange measurements with an alternating three-site mode ». Les trois sites, disposés en cercle, subissent des changements successifs de conformation (nous verrons plus loin comment). L’apport énergétique du flux de protons permet la fixation d’ADP+Pi sur un site et le relargage d’ATP fortement fixé sur un autre site.
Le modèle expérimental de la bactériorhodopsine
Halobacterium est une Archée vivant dans les milieux à forte concentration en sels. Elle tire son énergie de la capture de photons solaires par la bactériorhopsine, une protéine de 26 kilodaltons comportant 7 hélices α transmembranaires. En 1971, Walther Stoeckenius et Dieter Oesterhelt (Université de Californie, San diego) montrèrent que la bactériorhodopsine est une pompe à protons. Elle possède un chromophore, le rétinal, qui confère à la membrane d’Halobacterium sa couleur pourpre. L’absorbtion d’un photon de couleur verte fait passer le rétinal à l’état excité (isomérisation cis/trans). La bactériorhopsine prend alors la conformation d’une pompe à protons active. Lorsqu’il est éclairé, Halobacterium halobium éjecte des protons dans le milieu, créant ainsi un gradient électrochimique pouvant atteindre 300 millivolts. Les protons éjectés reviennent dans la cellule en empruntant un canal traversant une ATP synthase membranaire. L’énergie libérée par le passage des protons est couplée à une synthèse d’ATP.
La bactériorhopsine forme, dans la membrane plasmique d’Halobacterium, des amas de molécules compactées en cristal bidimensionnel. Cet arrangement est suffisamment ordonné pour permettre l’étude de la structure cristallographique aux rayons X. L’étude d’Halobacterium halobium a fourni des éclaircissements sur le mécanisme de conservation de l’énergie couplé à un transport unidirectionnel de protons, à travers une membrane, contre un gradient électrostatique. Les mécanismes chez Halobacterium et dans la membrane mitochondriale interne présentent des différences : (i) dans le cas de la bactériorhopsine l’énergie nécessaire au transfert de protons est fournie par l’isomérisation cis/trans du rétinal ayant absorbé un photon ; pour les oxydoréductases mitochondriales, l’énergie est libérée par les chûtes de potentiels entre deux transporteurs d’électrons. Les transporteurs sont assemblés dans la chaîne respiratoire par ordre de potentiel redox décroissant. (ii) La bactériorhodopsine est une molécule de taille modeste (26 kilodaltons) ; les oxydoréductases mitochondriales sont des complexes de grande taille : 240 kilodaltons pour le monomère du complexe des cytochromes bc1 ou de la cytochrome oxydase. En dépit de ces différences, le mécanisme du pompage des protons reste essentiellement le même. Dans la bactériorhodopsine, les résidus d’acides aminés impliqués ont été identifiés : deux aspartates, deux glutamates, une arginine et une base de Schiff au niveau du rétinal. Ils oscillent de l’état protoné à l’état déprotoné, véhiculant ainsi le proton d’un résidu à l’autre, selon une séquence déterminée. Dans les complexes I et IV, comme pour la bactériorhodopsine, le pompage unidirectionnel de protons implique des changements de conformations.
« The Fundamental Particles of Biology »
En 1961, Takuzo Oda et David E. Green (Institute for Enzyme Research, University of Wisconsin) demandèrent à Humberto Fernandez-Moran (Mixter Laboratories for Electron Microscopy, Massachusetts General Hospital, Boston) d’examiner au microscope électronique des membranes internes de mitochondries de cœur de bœuf colorées négativement à l’acide phosphotungstique. Les clichés révélèrent la présence de particules élémentaires (inner membrane spheres) de 9 à 10 nm de diamètre, rattachées à la membrane par une courte tige. Cette observation fut confirmée par D. F. Parsons et par Walther Stoeckenius (The Rockefeller University). Des particules semblables furent observées dans les membranes de chloroplastes et chez Escherichia coli. Green supposa qu’elles représentaient les unités, jusque-là hypothétiques, du système mitochondrial de transduction de l’énergie ; pour souligner leur importance, il les baptisa « The Fundamental Particles of Biology ». L’élucidation de leur structure atomique tridimensionnelle et de leur mode de fonctionnement représente une avancée majeure en biologie. Parmi ceux qui y contribuèrent, je placerai sur le devant de la scène les groupes d’Efraïm Racker (Biochemistry Department, Cornell University), Peter Mitchell (Glynn Research Ltd, Bodmin, Cornwall), Paul D. Boyer (Molecular Biology Institute, University of California, Los Angeles), et John E. Walker (Medical Research Council, Laboratory of Molecular Biology, Cambridge). Mitchell reçut le prix Nobel de chimie en 1978, Boyer et Walker furent colauréats de ce prix en 1997. Malgré l’importance de l’ensemble ses contributions (dont la découverte de la voie des pentoses phosphate), Racker ne figure pas parmi les lauréats ; faut-il y voir une conséquence de son retard à détecter les fraudes scientifiques de son collaborateur, Max Spector ?
Références: Fernandez-Moran H, Oda T, Blair PV, Green DE A macromolecular repeating unit of mitochondrial structure and function. Correlated electron microscopic and biochemical studies of isolated mitochondria and submitochondrial particles of beef heart muscle (1964)
Efraïm Racker et Yasuo Kagawa firent le rapprochement entre les particules de Fernandez-Moran et l’enzyme purifié en 1960 par Maynard E. Pullman et coll ; après traitement de la membrane mitochondriale interne par des protéases, ils avaient isolé une ATPase. Les protéases avaient coupé le lien réunissant le complexe enzymatique F1 (Fraction 1), au domaine Fo (Fraction oligomycine ; un antibiotique inhibant l’ATP synthase) intégré dans la membrane mitochondriale interne. Depuis les travaux de Pullman, la fonction du domaine F1 a été élucidée : sous la pression du potentiel de membrane provoqué par l’accumulation de protons pompés dans l’espace inter-membranaire par les complexes de la chaîne respiratoire, le domaine F1 laisse échapper des protons vers la matrice et fonctionne comme une synthase, catalysant la phosphorylation d’ADP en ATP. Dans le cas d’une inversion de la force proton-motrice, le domaine F1 fonctionnerait comme une ATPase, pompant des protons avec consommation d’ATP, comme c’est le cas chez les organismes chimiotrophes. Une étape de solubilisation par le cholate (un sel d’acide biliaire) fut nécessaire pour libérer le domaine polaire Fo de la membrane mitochondriale interne ; sa structure partielle fut établie par Yasuo Kagawa et Racker en 1966 ; ce domaine est formé d’un anneau de sous-unités c (10 à 15).
Références: Pullman ME, Penefsky HS, Datta A, Racker E Partial Resolution of the Enzymes Catalyzing Oxidative Phosphorylation. I. Purification and Properties of Soluble, Dinitrophenol-stimulated Adenosine Triphosphatase (1960)
Kagawa Y, Racker E Partial Resolution of the Enzymes Catalyzing Oxidative Phosphorylation. X. Correlation of morphology and function in submitochondrial particles (1966)
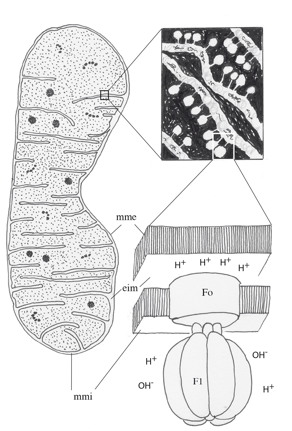 |
| Particules élémentaires. Après gonflement des mitochondries dans une solution hypotonique et coloration négative au phosphotungstate, les particules élémentaires, ou sphères pédonculées, sont visibles au microscope électronique. Elles sont reliées à la membrane interne par une tige. La partie sphérique, en contact avec la matrice, est le domaine catalytique F1 de l’ATP synthase. Les sphères pédonculées représentent 15 % de la masse des protéines de la membrane mitochondriale. |
Les données expérimentales, notamment de microscopie électronique, avaient révélé que l’ATP synthase F1Fo est composée d’une partie F1 faisant saillie dans la matrice mitochondriale et contenant les sites catalytiques, et d’une partie Fo, membranaire, contenant le canal à protons. L’ATPase F1Fo de mitochondries de cœur de bœuf fut purifiée par Alan E. Senior (Department of Biochemistry and Biophysics, University of Rochester Medical Center) et Harvey S. Penefsky (Department of Biochemistry and Molecular Biology, State University of New York, Syracuse); cinq polypeptides furent caractérisés : a (55.250 daltons), b (51.700), g (30.140), d (15.000), e (5.630). Les premières images de ce moteur nano-moléculaire, qui fournit l’essentiel des besoins en ATP de la cellule, furent obtenues dans le groupe d’Egbert Boekema (Rijksuniversiteit Groningen, Biomolecular Sciences and Biotechnology Institute) au prix d’un fastidieux travail d’analyse statistique des données de centaines de clichés de microscopie électronique de particules colorées négativement. Boekema et coll. obtinrent ultérieurement une représentation tridimensionnelle de l’ATPase F1Fo en utilisant la cryo-microscopie électronique d’échantillons congelés dans l’azote ou l’hélium liquides, combinée à la tomographie.
Références : Senior AE, Brooks JC Studies on the mitochondrial oligomycin-insensitive ATPase. I. An improved method of purification and the behavior of the enzyme in solutions of various depolymerizing agents (1970)
Brooks JC, Senior AE Methods for purification of each subunit of the mitochondrial oligomycin-insensitive adenosine triphosphatase (1970)
Penefsky HS Preparation of beef heart mitochondrial ATPase (1979)
Boekema E, Berden JA, van Heel M Structure of mitochondrial F1-ATPase studied by electron microscopy and image processing (1986)
Jan Pieter Abrahams et coll. établirent la structure cristallographique de la F1-ATPase de mitochondries de bœuf à une résolution de 2.8 Å. Les trois sous-unités catalytiques β ont des conformations et un contenu en nucléotide différents. Ces observations sont en accord avec un modèle catalytique de l’ATP synthase dans lequel les trois sous-unités β changent de conformation à chaque étape du cycle catalytique. Le passage d’une conformation à l’autre pourrait être provoqué par la rotation du bloc α3 β3 autour du domaine en hélice α de la sous-unité γ. Les biochimistes avaient montré que les parties F1 et Fo sont reliées par un filament, long de 45 Å, constitué des sous-unités γ et ϵ. Les sous-unités δ et b participent aussi à cette liaison. En 1997, Stephen Wilkens et coll. déterminèrent la structure en hélices α d’un fragment de la sous-unité δ, par spectroscopie par résonance magnétique nucléaire. Les premières structures cristallographiques obtenues par diffraction des rayons X dans le groupe de John E. Walker (Medical Research Council Laboratory of Molecular Biology, Cambridge), et dans d’autres laboratoires, ne concernaient que des fragments de la partie catalytique (F1-ATPase) : sous-complexes a3b3gde-c10 de Saccharomyces cerevisiae (Stock et coll.) ; a3b3gde de mitochondries de cœur de bœuf (Gibbons et coll.), a3b3g de chloroplastes d’épinards (Groth et Pohl). Une structure cristallographique de l’ATPase F1Fo partiel (7 chaînes polypeptidiques, 22.722 atomes) de Saccharomyces cerevisiae fut publiée en 1999. La structure de l’ATPase F1, associée à une protéine régulatrice inhibant l’activité ATPase, fut réalisée par Elena Cabezon en 2003.
Références : Abrahams P, Leslie AG, Lutter R, Walker JE Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria (1994)
Wilkens S, Rodgers A, Ogilvie I, Capaldi RA Structure and arrangement of the δ subunit in the E. coli ATP synthase (ECF1Fo) (1997)
Stock D, Leslie AG, Walker JE (1999) Molecular architecture of the rotary motor in ATP synthase (1999)
Gibbons C, Montgomery MG, Leslie AG, Walker JE (2000) The Structure of the central stalk in bovine F1-ATPase at 2.4 A resolution (2000)
Groth G, Pohl E The structure of the chloroplast F1-ATPase at 3.2 A resolution (2001)
Cabezon E, Montgomery MG, Leslie AGWL, Walker JE The structure of bovine F1-ATPase complexed with its regulatory protein, IF1 (2003)
En réunissant les données obtenues par l’analyse de deux structures tridimensionnelles, John L. Rubinstein, dans le laboratoire de John E. Walker, utilisa la cryo-microscopie électronique de particules isolées de mitochondries de coeur de bœuf pour déterminer la disposition des différents composants au sein de l’ATPase F1, à une résolution de 32 Å. Le sous-complexe F1-c10 contient les tige centrale et périphérique ; le sous-complexe Fo est composé de deux domaines : l’un correspondant à l’anneau des sous-unités c et l’autre, à la sous-unité a, la portion transmembranaire de la sous-unité b et des protéines membranaires intégrales. La flexibilité de la tige périphérique selon qu’elle interagit ou pas avec la partie α–β de F1 suggère qu’elle pourrait jouer le rôle de stator au cours de la synthèse ou de l’hydrolyse de l’ATP. John E. Walker reçut le prix Nobel de chimie 1997.
Référence : Rubinstein JL, Walker JE, Henderson R Structure of the mitochondrial ATP synthase by electron cryomicroscopy (2003)
La notion que les nano-moteurs rotatoires existent dans la membrane sous forme de super-complexes émergea avec la mise à la disposition des chercheurs de l’électrophorèse sur gel de polyacrylamide en conditions non dénaturantes – natives (blue-native polyacrylamide gel electrophoresis, « native PAGE »). On a établi, par examen au microscope électronique de particules isolées, la structure des complexes I monomérique et II dimérique d’Arabidopsis, et du complexe dimérique de l’algue Polytomella. Pour Alexander Hahn et coll. et Karen M. Davies et coll. (Department of Structural Biology, Max Planck Institute of Biophysics, Frankfurt am Main), la courbure de la membrane mitochondriale interne (cristae) au niveau des dimères d’ATP synthase est imposée par la pliure particulière de la portion membranaire de l’ATP synthase.
Références : Hahn A, Parey K, Bublitz M, Mills DJ, Zickermann V, Vonck J, Kühlbrandt W, Meier T Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology (2006)
Davies KM, Anselmi C, Wittig I, Kühlbrandt Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae (2012)
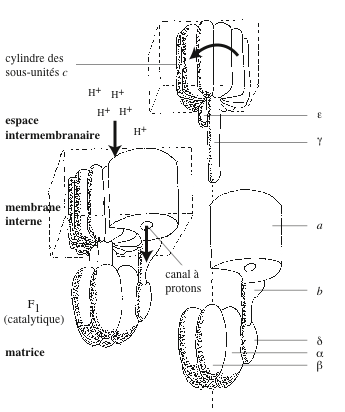 |
| ATP synthase. Ce convertisseur d’énergie est formé de deux moteurs. Le moteur Fo comporte une partie fixe composée des sous-unités membranaires: a (traversée par le canal à protons), 2 b et δ ; et une partie mobile constituée d’un anneau de sous-unités c (∼10 copies) inséré dans la membrane interne. Le moteur F1 comporte les sous-unités γ (un cylindre de 16 nanomètres de long), ε et l’hexamère 3α 3β, une sphère aplatie de 8 x 10 nm. Les sous-unités β renferment le site catalytique où s’opère la conversion d’ADP en ATP. |
La théorie chimio-osmotique formulée par Peter Mitchell en 1961 repose sur l’existence d’un gradient électrochimique de protons de part et d’autre de la membrane mitochondriale interne. Les chutes de potentiel entre complexes transporteurs d’électrons de la membrane mitochondriale interne actionnent le pompage de protons dans l’espace inter-membranaire ; d’autre part, à la différence de la membrane interne, imperméable aux protons (elle est riche en cardiolipines), la membrane mitochondriale externe est perméable aux protons libérés dans le cytoplasme par le métabolisme intermédiaire ; leur accumulation dans l’espace inter-membranaire crée un gradient électrochimique dû à l’addition de deux facteurs : (i) la polarité de la membrane : la face en contact avec l’espace inter-membranaire est négative ; celle tournée vers la matrice est positive (différence de charge : environ 80 mV) ; (ii) la concentration en H+ dans l’espace inter-membranaire par rapport à la matrice (environ 2 unités de pH). L’hypothèse de Mitchell implique que pour diffuser de l’espace inter-membranaire vers la matrice, les ions H+ traversent l’ATP synthase. D’abord rejetée par ses pairs, l’hypothèse de Mitchell reçut une première validation expérimentale lorsque John E. Walker et ses collaborateurs du Laboratory of Molecular Biology à Cambridge, établirent la structure atomique tridimensionnelle de l’ATP synthase à une résolution de 2.8 Å. En 1999, ce fut le tour de la structure de l’ATP synthase de Saccharomyces cerevisiae, un assemblage de 13 sous unités (masse moléculaire : environ 600 000 daltons) réparties en domaines structurels et fonctionnels : la F1ATPase comporte une tête globulaire catalytique α(3) β(3) traversée par la tige centrale asymétrique γ(1) ; la Fo ATPase est formée principalement de protéines membranaires ; elle comporte en particulier le domaine a, solidaire de la tige périphérique b(2), δ(1), en contact avec les protomères α et β du domaine F1 et orientée vers la matrice mitochondriale, le stroma des chloroplastes ou le cytosol des bactéries. L’ATP synthase est un moteur moléculaire composé d’un stator, avec les sous-unités a(1) de Fo, b(2), δ(1), α(3) β(3) de F1, et un rotor composé de l’anneau des sous-unités c(9-12) et de la tige γ(1), δ(1), ε(1).
Un autre résultat expérimental étaya de façon convaincante l’hypothèse de Mitchell ; il fut obtenu par André T. Jagendorf dans le laboratoire d’Efraïm Racker, à Cornell University (voir « Hypothèse chimio-osmotique ») ; en 1966, Jagendorf et Uribe ont mis en évidence une synthèse d’ATP in vitro par des chloroplastes chargés en protons.
Références: Abrahams JP, Leslie AG, Lutter R, Walker JE Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria (1994)
Stock D, Leslie AGW, Walker JE Molecular Architecture of the Rotary Motor in ATP Synthase (1999)
Dickson VK, Silvester JA, Fearnley IM, Leslie AG, Walker JE On the structure of the stator of the mitochondrial ATP synthase (2006)
Rees DM, Leslie AG, Walker JE The structure of the membrane extrinsic region of bovine ATP synthase (2009)
Rubinstein JL, Walker JE, Henderson R Structure of the mitochondrial ATP synthase by electron cryomicroscopy (2003)
Lau WC, Baker LA, Rubinstein JL Cryo-EM structure of the yeast ATP synthase (2008)
« A splendid molecular machine »
En 1974, Paul D. Boyer proposa la théorie du mécanisme de changement de liaison (Binding Change Mechanism), plus connue sous l’appellation de catalyse rotationnelle. Les trois postulats qu’il avança pour expliquer le fonctionnement de l’ATP synthase furent validés par la mise en évidence de la « turbine moléculaire rotatoire » qui, chez les mitochondries, les chloroplastes et certaines bactéries, est au cœur du système de conversion d’énergie osmotique en énergie chimique. Le fonctionnement de l’ATP synthase est déclenché par la rotation du cylindre des sous-unités c, entraînant dans son mouvement la sous-unité assymétrique γ. Les polypeptides du cylindre c, enchâssé dans la membrane, comportent deux hélices α constituées d’acides aminés hydrophobes à l’exception d’un résidus aspartate (Asp) en position 61. L’ensemble des Asp 61 forme un anneau situé vers le milieu de la bicouche phospholipidique, ce qui empêche la rotation spontanée du cylindre. Lorsqu’un proton traverse le canal (en réalité les deux demi-canaux) de la sous-unité a, il neutralise la charge d’un Asp 61. L’aspartate protoné entre en contact avec la bicouche hydrophobe et la rotation de l’anneau c est enclenchée entrainant la sous-unité γ qui lui est attachée ; celle-ci pénètre dans l’hexamère α(3) β(3) maintenu en position fixe par l’ensemble des sous-unités du stator, l’empêchant ainsi de suivre le mouvement de rotation. Il se produit une conversion d’énergie osmotique du gradient électrochimique en énergie mécanique de transconformation des sous-unités β ; elles passent par trois conformations qui se succèdent dans l’ordre : lâche (fixation d’ADP + Pi), serrée (condensation en ATP), ouverte (libération de l’ATP) ; il y a en permanence une sous-unité β dans chacune de ces trois conformations et une molécule d’ATP est synthétisée à chaque rotation du rotor de 120° (3 ATP par rotation de 360°). De l’énergie mécanique est consommée pour charger en énergie chimique une molécule d’ATP, le seul organophosphate rechargeable par couplage chimio-osmotique. Dans les conditions qui prévalent dans la cellule, la variation d’enthalpie libre lors de l’hydrolyse de l’ATP en ADP est ΔG = -51,8 kJ/mole
En 1997, Hiroyuki Noji, Ryohei Yasuda, Masasuke Yoshida et Kazuhiko Kinoshita Jr. (Research Laboratory of Resources Utilization, Tokyo Institute of Technology, Yokohama) firent la démonstration visuelle directe que la F1-ATPase est un moteur moléculaire rotatoire – comme l’énorme complexe (plus de 100 polypeptides) du flagelle bactérien. Ils fixèrent sur une surface plane recouverte d’une couche de nickel les sous-unités β du sphéroïde α(3) β(3) par une queue poly-histidine. Ils attachèrent par une liaison covalente un long filament d’actine fluorescente à la sous-unité γ émergent du sphéroïde. Les mouvements rotatoires du bras d’actine purent être directement observés au microscope à fluorescence. On estime que le rotor effectue 130 révolutions par seconde.
Références: Dahms AS, Boyer PD Occurrence and Characterisation of 18O-exchange Reactions Catalyzed by Sodium-and Potassium-dependent Adenosine Triphosphatases (1972)
Boyer PD The ATP synthase–a splendid molecular machine (1997)
Noji H, Yasuda R, Yoshida M, Kinoshita Jr. K Direct observation of the rotation of F1-ATPase (1997)
Boyer PD What makes ATP synthase spin ? (1999)
Schnitzer MJ Doing a rotary two-step (2001)
Bilan
Etablissons le bilan énergétique en partant de l’oxydation du glucose. Après passage par la glycolyse (+ 2 ATP), le cycle de Krebs (+ 2 ATP) et la chaîne respiratoire (+ 34 ATP) une molécule de glucose produit théoriquement 38 molécules d’ATP. Le bilan doit être minoré (30 à 32 molécules d’ATP) pour tenir compte du coût énergétique du transport de certaines molécules du cytosol vers la matrice mitochondriale (pyruvate).
Cytochrome c mitochondrial et apoptose
Sous ses différentes formes, la nécrose cellulaire et tissulaire est un phénomène connu de longue date. Elle est le plus souvent consécutive à un épisode traumatique et se traduit par le gonflement et l’éclatement des cellules. Le cytoplasme répandu affecte les cellules voisines, ce qui entraîne une inflammation et une réaction immunitaire. L’apoptose fut identifiée comme un phénomène de mort cellulaire différent de la nécrose à la suite des travaux des embryologistes et des pathologistes. En 1951, le biologiste Alfred Glücksmann (Strangeways Research Laboratory, Cambridge) fit paraître un article de revue dans les Biological Reviews of the Cambridge Philosophy Society. La diffusion de ce journal étant restreinte, cet article passa largement inaperçu aux yeux des non-spécialistes de la biologie du développement des vertébrés. Glücksmann y proposait l’idée qu’au même titre que la mitose et à l’opposé, la mort cellulaire est un mécanisme naturel de régulation des populations cellulaires, à l’origine de la formation des tissus et des organes.
Référence : Glücksmann A Cell Deaths in Normal Vertebrate Ontogeny (1951)
Dix ans plus tard, le phénomène de mort cellulaire programmée allait réapparaître comme une conclusion des travaux des pathologistes sur les dommages infligés à des tissus soumis à différentes formes de stress : ischémie (John Foxton R. Kerr, University College, Hospital Medical School, London), traitement par un hydrocarbure polycyclique cancérigène (Alastair R. Currie, University of Aberdeen, Ecosse), sevrage en facteur de croissance (Andrew H. Wyllie, University of Aberdeen). L’examen au microscope optique ou électronique révéla la présence d’agrégats de chromatine dans le noyau et d’une fragmentation du cytoplasme avec formation de corps apoptotiques, des vésicules limitées par une membrane et contenant des organites. Les corps apoptotiques sont immédiatement phagocytés par les cellules voisines ; ils subissent une ségrégation dans les phagosomes, puis une digestion après fusion des phagosomes avec des lysosomes.
John F.R. Kerr (University of Queensland, Brisbane, Australia) décrivit, en 1965, ce qu’il appelait « shrinkage necrosis » dans un article publié dans le Journal of Pathology and Bacteriology. En 1972, après avoir comparé leurs résultats avec ceux des embryologistes, John Kerr, son étudiant post-doctoral Alastair Currie et Andrew Wyllie avancèrent l’hypothèse de mort cellulaire programmée dans un article publié dans le British Journal of Cancer, dans lequel ils adoptèrent le terme « apoptose » qui leur avait été suggéré par James Cormack (Department of Greek, University of Aberdeen).
Références : Kerr JFR A Histochemical Study of Hypertrophy and Ischaemic Injury of Rat Liver with Special Reference to Changes in Lysosome (1965)
Kerr JFR, Currie A, Wyllie A Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics (1972)
Pendant une décennie, le mécanisme de l’apoptose à l’échelle moléculaire ne suscita aucun intérêt de la part des biochimistes ; ce n’est qu’à partir des années 1980 qu’un certain nombre d’entre eux commencèrent à s’y intéresser. Selon l’origine des signaux, intracellulaires ou extracellulaires, qui déclenchent l’apoptose, on distingue deux grandes voies de signalisation : la voie extrinsèque, initiée par des signaux extérieurs à la cellule, fait intervenir des récepteurs de la membrane plasmique : (i) Tumor Necrosis Factor Receptor 1, qui fixe le Facteur de nécrose tumorale : Tumor Necrosis Factor α. (ii) le récepteur Fas (Fas Receptor), qui fixe le ligand membranaire Fas Ligand lié à la surface de cellules du système immunitaire) (iii) Les Death Receptors DR4 et DR5, qui fixent le ligand TRAIL (Tumor necrosis factor-associated ligand inducing apoptosis). La liaison des ligands à leur récepteurs aboutit in fine à l’activation d’enzymes protéolytiques à cystéine, les caspases (Cysteine ASPartic proteASEs). Étudiées depuis la découverte de l’Interleukin 1 Converting Enzyme (caspase 1), dans les années 1990, ces protéases sont présentes dans le cytosol sous forme de proenzymes inactifs. L’association ligands – récepteurs entraîne la formation du complexe intracellulaire DISC (Death-Inducing Signaling Complex) qui active la cascade des caspases, d’abord la caspase initiatrice 8, puis les caspases effectrices de l’apoptose. Dans le mécanisme que je viens de décrire brièvement, il faut souligner que l’internalisation rapide des récepteurs, déclenchée par la fixation des ligands, joue un rôle essentiel dans le déclenchement de l’apoptose.
Références : Artykov AA, Yagolovitch AV, Dolgikh DA, Kirpichnikov MP, Trushina DB, Gasparian ME Death Receptors DR4 and DR5 Undergo Spontaneous and Ligand-Mediated Endocytosis and Recycling Regardless of the Sensitivity of Cancer Cells to TRAIL (2020)
Schütze S, Schneider-Bracher W Impact of TNF-R1 and CD95 internalization on apoptotic and antiapoptotic signaling (2009)
La voie intrinsèque (voie mitochondriale), le principal mode d’activation de l’apoptose, met en jeu des protéines de la super-famille Bcl-2 (B Cell Lymphoma 2). Ces protéines dimériques occupent une position centrale dans la régulation de l’apoptose : le dimère Bcl-2-Bcl-2 exerce une activité anti-apoptotique tandis que les dimères de Bax (Bcl-2 associated X) ou de Bak sont pro-apoptotiques. La voie intrinsèque est le mode d’activation de l’apoptose pendant le développement embryonnaire ; cette voie est aussi activée par des signaux venant de la cellule, comme le stress oxydatif ou des dommages infligés à l’ADN et qui ne peuvent être corrigés par les systèmes de réparation. La protéine cytosolique Bax s’unit à elle-même et l’homodimère Bax-Bax se fixe sur la membrane mitochondriale externe, provoquant la formation de méga-pores (Mitochondrial Permeability Transition Pores). Le cytochrome c et une flavoprotéine : le Facteur inducteur d’apoptose (Apoptosis-Inducing Factor, AIF), localisés dans l’espace inter-menbranaire mitochondrial, sont libérés dans le cytosol et se lient l’un à l’autre. Cette association favorise le recrutement d’autres molécules d’AIF pour former un complexe en forme d’anneau appelé « apoptosome ». L’interaction du domaine CARD (Caspase Apaf 1 Recruitment Domain) d’Apaf 1 avec le domaine CARD amino-terminal du pro-enzyme de la caspase-9, en présence d’ATP, déclenche le recrutement et l’activation de la protéase (dans la voie extrinsèque, c’est la procaspase-8 qui est recrutée par les ligands des récepteurs du TNF), qui déclenche à son tour la cascade des caspases effectrices (caspases 3 et 7) responsables de la digestion des protéines de structure, de la fragmentation du cytosquelette, de la dégradation de l’ADN nucléaire et du démantèlement final de la cellule.
Référence : Zhou M, Li Y, Hu Q, Bai XC, Huang W, Yan C, Sheres SHW, Shi Y Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1 (2015)
Pathologie mitochondriale
Avant d’aborder ce thème, il me semble utile de rappeler que les protéines mitochondriales sont codées par l’ADN nucléaire et par l’ADN mitochondrial. L’ADNmt est transmis selon les lois de l’hérédité maternelle. Chez l’homme, il est constitué de 16.569 paires bases ; 37 gènes codent pour 22 ARNt, 2 ARNr, et 13 protéines des complexes de la chaîne respiratoire: les sous-unités 1, 2, 3, 4, 4L, 5 et 6 du complexe I, le cytochrome b du complexe III, les sous-unités 2 et 3 du complexe IV, et les sous-unités 6 et 8 du complexe V. L’ADN nucléaire est constitué de trois milliards de paires bases ; il code pour la majorité des protéines mitochondriales. Les maladies mitochondriales résultent de mutations touchant soit l’ADNmt, soit l’ADN nucléaire. Les pathologies mitochondriales primaires affectent les organes les plus gourmands en énergie ; les dysfonctionnements mitochondriaux secondaires – stress oxydatif, perte de contrôle de la qualité mitochondriale – se manifestent dans les maladies liées au vieillissement – Alzheimer, Parkinson – ou dans les maladies cardiovasculaires.
Article de revue : Fetterman JF, Chinery PF, McClellan, Wallace DC, Suomalainen A, Oala T, Lewis SC, Ballinger SW Mitochondrial Genetics in Cardiovascular Health and Disease: A Scientific Statement From the American Heart Association (2025)
Lorsque l’on aborde le domaine des pathologies mitochondriales, il faut avoir présent à l’esprit le concept d’hétéroplasmie, qui explique la très grande hétérogénéité des symptômes cliniques chez les patients. On l’explique ainsi : les cellules contiennent de nombreuses mitochondries (environ 2.000 dans un hépatocyte) possédant chacune 2 à 10 copies d’ADNmt. Une mutation peut n’affecter qu’un nombre restreint de copies. Le seuil à partir duquel un déficit en énergie entraîne des symptômes varie fortement selon les tissus (cerveau, muscles, cœur, et les systèmes visuel et auditif). Le système nerveux central étant un grand consommateur d’énergie, il est affecté au premier chef par les dysfonctionnements de la chaîne respiratoire. C’est ainsi que le Syndrome MELAS ou le Syndrome de Leigh sont des affections neurologiques graves, au pronostic sévère, qui affectent les très jeunes enfants ou les jeunes adultes. Dans le Syndrome MELAS, le déficit porte sur le fonctionnement des complexes I et/ou IV. Sur le plan clinique, ce syndrome se traduit par une myopathie affectant les fibres musculaires rouges, une encéphalopathie et de l’acidose lactique (le fonctionnement déficient de la chaîne respiratoire provoque une conversion de pyruvate en lactate). La plupart des patients présentent une mutation du gène codant l’ARNtLeu.
Le bras long du chromosome 9 contient un amas très dense de gènes, le Surfeit gene cluster, qui regroupe six gènes (SURF-1 à 6) étroitement associés. Le produit du gène SURF1, codé par l’ADN nucléaire, est adressé à la membrane mitochondriale interne. La protéine SURF-1 est un facteur indispensable à l’assemblage des sous-unités du complexe IV de la chaîne respiratoire (cytochrome c oxydase). Certains individus portent une mutation homozygote du gène SURF-1 via une transmission génétique autosomique récessive (le gène muté est transmis par les deux parents). Le syndrome de Leigh (encéphalomyélopathie nécrosante subaiguë) est une maladie neurodégénérative sévère, qui résulte du défaut d’assemblage du complexe IV ; elle apparaît chez le nourrisson ou le jeune enfant, entraînant un retard de développement, une faiblesse musculaire, des lésions dans des zones spécifiques du cerveau et des problèmes respiratoires.
Article de revue : Mei J, Ding P, Gao C, Zhou J, Li Z, Zhang C, Gao J Mitochondrial Diseases: Molecular Pathogenesis and Therapeutic Advances (2025)